vaccin oral à rotavirus (vivant)
FORMES et PRÉSENTATIONS
|
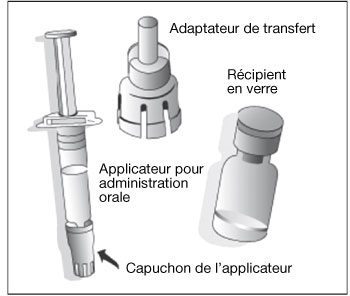
Poudre (blanche) et solvant (liquide trouble avec un surnageant incolore et un dépôt blanc sédimentant lentement) pour suspension buvable : Récipient de poudre + 1 applicateur pour administration orale de 1 ml de solvant + 1 adaptateur de transfert.
COMPOSITION
|
| p dose de 1 ml* | ||
| Rotavirus humain, souche RIX4414 (vivante atténuée)** |
>= 106.0 DICC50 | |
Excipients :
Poudre : saccharose, dextran, sorbitol, acides aminés, milieu Eagle modifié de Dulbecco (DMEM).
Solvant : carbonate de calcium, gomme xanthane, eau stérile.
Teneur en saccharose : 9 mg/ml.
Teneur en sorbitol : 13,5 mg/ml.
*
après reconstitution
**
produite sur cellules Vero
INDICATIONS
|
Rotarix est indiqué dans l’immunisation active des nourrissons à partir de l’âge de 6 semaines pour la prévention des gastroentérites dues à une infection à rotavirus (cf Posologie/Mode d’administration).
POSOLOGIE ET MODE D’ADMINISTRATION
|
Posologie :
Le schéma de vaccination comporte 2 doses. La première dose peut être administrée à partir de l’âge de 6 semaines. L’intervalle entre les doses doit être au moins de 4 semaines. Le schéma de vaccination doit préférentiellement être administré avant l’âge de 16 semaines, et doit être terminé avant l’âge de 24 semaines.
Rotarix peut être administré à la même posologie à des nourrissons nés prématurés à 27 semaines de grossesse ou plus (cf Effets indésirables et Pharmacodynamie).
Dans les essais cliniques, le vaccin a rarement été craché ou régurgité et, dans ces circonstances, une dose de remplacement n’a pas été administrée. Cependant, dans l’éventualité rare où un nourrisson recrache ou régurgite la majeure partie de la dose administrée de vaccin, une dose unique de remplacement peut être donnée lors de la même consultation.
Il est recommandé aux nourrissons qui ont reçu une première dose de Rotarix de terminer le schéma de vaccination en 2 doses avec Rotarix. Il n’y a aucune donnée de tolérance, d’immunogénicité ou d’efficacité quand Rotarix est administré comme première dose et un autre vaccin à rotavirus comme seconde dose ou vice versa.
Mode d’administration :
Rotarix doit être administré uniquement par voie orale.
Rotarix ne doit jamais être injecté.
CONTRE-INDICATIONS
|
- Hypersensibilité à la substance active ou à l’un des excipients.
- Hypersensibilité suite à une précédente administration de vaccins rotavirus.
- Antécédents d’invagination intestinale.
- Sujets ayant une malformation congénitale non opérée du tractus gastro-intestinal pouvant prédisposer à une invagination intestinale.
- L’administration de Rotarix doit être différée chez les sujets ayant une maladie fébrile sévère aiguë. La présence d’une infection bénigne n’est pas une contre-indication à la vaccination.
- L’administration de Rotarix doit être différée chez les sujets présentant une diarrhée ou des vomissements.
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI
|
La vaccination doit être précédée d’une recherche des antécédents médicaux, notamment concernant les contre-indications, et d’un examen clinique.
INTERACTIONS
|
Rotarix peut être administré simultanément avec l’un des vaccins monovalents ou combinés suivants (incluant les vaccins hexavalents [DTCa-HepB-IPV/Hib]) : vaccin diphtérie-tétanos-coqueluche à germes entiers (DTCe), vaccin diphtérie-tétanos-coqueluche acellulaire (DTCa), vaccin Haemophilus influenzae de type b (Hib), vaccin poliomyélitique inactivé (IPV), vaccin hépatite B (HepB), vaccin conjugué pneumococcique et vaccin conjugué méningococcique du groupe C. Les études cliniques ont montré que les réponses immunitaires et les profils de tolérance des vaccins administrés n’étaient pas modifiés.
GROSSESSE et ALLAITEMENT
|
Rotarix n’étant pas destiné à l’adulte, les données concernant l’utilisation du vaccin chez la femme pendant la grossesse ou l’allaitement et les études de reproduction chez l’animal ne sont pas disponibles.
EFFETS INDÉSIRABLES
|
-
Essais cliniques : - Le profil de sécurité présenté ci-dessous repose sur les données issues des essais cliniques réalisés, soit avec la formulation lyophilisée, soit avec la formulation liquide de Rotarix.
- Au cours de 4 essais cliniques, environ 3800 doses de Rotarix formulation liquide ont été administrées à environ 1900 nourrissons. Ces essais ont montré que le profil de sécurité de la formulation liquide est comparable à celui de la formulation lyophilisée.
- Au cours des 23 études cliniques, environ 106 000 doses de Rotarix (formulation lyophilisée ou liquide) ont été administrées à environ 51 000 nourrissons.
- Dans 3 essais cliniques contrôlés versus placebo (Finlande, Inde et Bangladesh), dans lesquels Rotarix a été administré seul (l’administration des vaccins pédiatriques usuels était décalée), l’incidence et la sévérité des événements, diarrhées, vomissements, perte d’appétit, fièvre, irritabilité et toux/écoulement nasal n’étaient pas significativement différentes dans le groupe recevant Rotarix comparé au groupe placebo. Aucune augmentation de l’incidence ou de la sévérité de ces événements n’a été observée après la seconde dose.
- Au cours d’une analyse poolée de 17 essais cliniques contrôlés versus placebo (Europe, Amérique du Nord, Amérique latine, Asie, Afrique) incluant les essais dans lesquels Rotarix était coadministré avec des vaccins pédiatriques habituels (cf Interactions), les effets indésirables suivants ont été considérés comme possiblement liés à la vaccination.
- Les réactions indésirables suivantes sont listées par classe-organe et fréquence.
- Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité.
- Les fréquences sont définies comme suit : très fréquent >= 10 %, fréquent >= 1 % et < 10 %, peu fréquent >= 0,1 % et < 1 %, rare >= 0,01 % et < 0,1 %.
- Affections gastro-intestinales :
- Fréquent : diarrhée.
- Peu fréquent : douleurs abdominales, flatulences.
- Affections de la peau et du tissu sous-cutané :
- Peu fréquent : dermatite.
- Troubles généraux et anomalies au site d’administration :
- Fréquent : irritabilité.
- Le risque d’invagination intestinale a été évalué dans une large étude clinique de tolérance, conduite en Amérique latine et en Finlande, dans laquelle 63 225 nourrissons étaient inclus. Cet essai ne montrait aucune augmentation du risque d’invaginations intestinales dans le groupe Rotarix par rapport au groupe placebo tel qu’indiqué dans le tableau ci-dessous.
-
Invaginations intestinales dans les 31 jours après l’administration de la Rotarix (N = 31 673) Placebo (N = 31 552) Risque relatif (IC* 95 %)
1re dose 1 2 0,50 (0,07 ; 3,80)
2e dose 5 5 0,99 (0,31 ; 3,21)
-
*
intervalle de confiance
-
Tolérance chez les nourrissons nés prématurés : - Dans une étude clinique, 670 nourrissons prématurés nés entre 27 et 36 semaines de grossesse ont reçu Rotarix et 339 un placebo. La première dose a été administrée à partir de l’âge de six semaines. Des événements indésirables graves ont été observés chez 5,1 % des nourrissons ayant reçu Rotarix et chez 6,8 % des nourrissons ayant reçu un placebo. Des taux similaires d’autres évènements indésirables ont été observés chez les nourrissons ayant reçu Rotarix ou un placebo. Aucun cas d’invagination intestinale n’a été rapporté.
-
Tolérance chez les nourrissons infectés par le virus de l’immunodéficience humaine (VIH) : - Dans une étude clinique, 100 nourrissons infectés par le VIH ont reçu Rotarix ou un placebo. Le profil de sécurité était similaire entre les nourrissons ayant reçu Rotarix et ceux ayant reçu le placebo.
-
Surveillance post-commercialisation : - Parce que ces événements sont issus de la notification spontanée, il n’est pas possible d’estimer de façon fiable leur fréquence.
- Affections respiratoires, thoraciques et médiastinales : apnée chez les grands prématurés (nés à 28 semaines de grossesse ou moins) ; cf Mises en garde/Précautions d’emploi.
- Affections gastro-intestinales : rectorragies.
- Dans les données post-commercialisation, des cas d’invagination intestinale ont été rapportés en association temporelle avec Rotarix. La plupart des cas ont été rapportés dans les 7 jours suivant la première dose (cf Mises en garde/Précautions d’emploi).
SURDOSAGE
|
Aucun cas de surdosage n’a été rapporté.
PHARMACODYNAMIE
|
Classe pharmacothérapeutique : vaccins contre les diarrhées à rotavirus, (code ATC : J07BH01).
-
Efficacité protectrice : - Des études cliniques ont été conduites en Europe et en Amérique latine pour évaluer l’efficacité protectrice de Rotarix contre toutes les gastroentérites et les gastroentérites sévères à rotavirus.
- Une étude clinique réalisée en Europe a évalué Rotarix administré selon différents schémas d’administration européens (2, 3 mois ; 2, 4 mois ; 3, 4 mois ; 3, 5 mois) chez 4000 sujets. La sévérité de la gastroentérite était définie selon l’échelle à 20 points de Vesikari qui évalue le tableau clinique complet de la gastroentérite à rotavirus en tenant compte de la sévérité et de la durée de la diarrhée et des vomissements, la sévérité de la fièvre et de la déshydratation ainsi que la nécessité de recourir à un traitement.
- Après 2 doses de Rotarix, l’efficacité protectrice du vaccin observée au cours des 1re et 2e années de vie est présentée dans le tableau ci-dessous :
-
1re année de vie
Rotarix N = 2572
Placebo N = 1302(a)2e année de vie
Rotarix N = 2554;
Placebo N = 1294(a)
Type Tout grade Sévère(b) Tout grade Sévère(b)
Efficacité du vaccin (%) contre les gastro-entérites à rotavirus de tout grade et les gastro-entérites à rotavirus sévères [IC 95%]
G1P[8] 95,6(c)
[87,9 ; 98,8]96,4(c)
[85,7 ; 99,6]82,7(c)
[67,8 ; 91,3]96,5(c)
[86,2 ; 99,6]
G2P[4] 62,0
[< 0,0 ; 94,4]74,7
[< 0,0 ; 99,6]57,1(c)
[< 0,0 ; 82,6]89,9(c)
[9,4 ; 99,8]
G3P[8] 89,9(c)
[9,5 ; 99,8]100,0(c)
[44,8 ; 100,0]79,7(c)
[< 0,0 ; 98,1]83,1
[< 0,0 ; 99,7]
G4P[8] 88,3(c)
[57,5 ; 97,9]100,0(c)
[64,9 ; 100,0]69,6
[< 0,0 ; 95,3]87,3(c)
[< 0,0 ; 99,7]
G9P[8] 75,6(c)
[51,1 ; 88,5)94,7(c)
[77,9 ; 99,4]70,5(c)
[50,7 ; 82,8]76,8(c)
[50,8 ; 89,7]
Souches contenant le génotype P[8] 88,2(c)
[80,8 ; 93,0]96,5(c)
[90,6 ; 99,1]75,7(c)
[65,0 ; 83,4]87,5(c)
[77,8 ; 93,4]
Souches de rotavirus circulantes 87,1(c)
[79,6 ; 92,1]95,8(c)
[89,6 ; 98,7]71,9(c)
[61,2 ; 79,8]85,6(c)
[75,8 ; 91,9]
Efficacité du vaccin (%) contre les gastro-entérites à rotavirus nécessitant une prise en charge médicale [IC 95%]
Souches de rotavirus circulantes 91,8(c)
[84,0 ; 96,3]76,2(c)
[63,0 ; 85,0]
Efficacité du vaccin (%) contre les hospitalisations causées par une gastro-entérite à rotavirus [IC 95%]
Souches de rotavirus circulantes 100(c)
[81,8 ; 100]92,2(c)
[65,6 ; 99,1]
-
(a)
Cohorte per protocole pour l’efficacité. -
(b)
Gastro-entérite sévère définie par un score >= 11 sur l’échelle de Vesikari. -
(c)
Statistiquement significatif (p < 0,05). - L’efficacité du vaccin durant la première année de vie a progressivement augmenté avec la sévérité de la maladie, en atteignant 100% (IC 95%: 84,7;100) pour les scores de Vesikari >= 17.
- Une étude clinique conduite en Amérique latine a évalué Rotarix chez plus de 20 000 sujets. La sévérité des gastroentérites était définie selon les critères OMS. L’efficacité protectrice du vaccin contre les gastroentérites sévères à rotavirus nécessitant une hospitalisation et/ou un traitement par réhydratation dans une structure médicale et l’efficacité spécifique du vaccin par sérotype après 2 doses de Rotarix sont présentés dans le tableau ci-dessous :
-
Sérotype Gastroentérite sévère à rotavirus
(1re année de vie)
Rotarix N = 9009 ; Placebo N = 8858(a)Gastroentérite sévère à rotavirus
(2e année de vie)
Rotarix N = 7175 ; Placebo N = 7062(a)
Efficacité (%)
[IC 95 %]Efficacité (%)
[IC 95 %]
Toute gastroentérite à rotavirus 84,7(b)
[71,7 ; 92,4]79,0(b)
[66,4 ; 87,4]
G1P[8] 91,8(b)
[74,1 ; 98,4]72,4(b)
[34,5 ; 89,9]
G3P[8] 87,7(b)
[8,3 ; 99,7]71,9
[< 0,0 ; 97,1]
G4P[8] 50,8(c)
[< 0,0 ; 99,2]63,1(b)
[0,7 ; 88,2]
G9P[8] 90,6(b)
[61,7 ; 98,9]87,7(b)
[72,9 ; 95,3]
Souches contenant le génotype P[8] 90,9(b)
[79,2 ; 96,8]79,5(b)
[67,0 ; 87,9]
-
(a)
Cohorte per protocole pour l’efficacité. -
(b)
Statistiquement significatif (p < 0,05). -
(c)
Le nombre de cas sur lequel repose l’estimation de l’efficacité contre le sérotype G4P[8] était faible (1 cas dans le groupe Rotarix et 2 cas dans le groupe placebo). - Une analyse poolée de 5 études d’efficacité* a estimé à 71,4 % (IC 95 % : 20,1 ; 91,1) l’efficacité contre les gastroentérites sévères à rotavirus (score Vesikari >= 11) dues au rotavirus de type G2P[4] pendant la 1re année de vie.
-
*
Dans ces études, l’efficacité estimée et les intervalles de confiance étaient respectivement : 100 % (IC 95% : – 1858,0 ; 100), 100 % (IC 95 % : 21,1 ; 100), 45,4 % (IC 95 % : – 81,5 ; 86,6) et 74,7 % (IC 95 % : – 386,2 ; 99,6). Aucune estimation n’était disponible dans la dernière étude.
-
Réponse immunitaire : - Le mécanisme immunologique selon lequel Rotarix protège contre les gastroentérites à rotavirus n’est pas complètement connu. La relation entre les réponses en anticorps après vaccination contre le rotavirus et la protection contre les gastroentérites à rotavirus n’a pas été établie.
- Le tableau suivant montre le pourcentage de sujets ayant un taux sérique en anticorps IgA anti-rotavirus >= 20 UI/ml (par ELISA) 1 à 2 mois après la seconde dose de vaccin ou placebo, dans les différentes études.
-
Schéma (mois) Études conduites en Vaccin Placebo
N % >= 20 UI/ml
[IC 95 %]N % >= 20 UI/ml
[IC 95 %]
2, 3 France, Allemagne 239 82,8
[77,5 ; 87,4]127 8,7
[4,4 ; 15,0]
2, 4 Espagne 186 85,5
[79,6 ; 90,2]89 12,4
[6,3 ; 21,0]
3, 5 Finlande, Italie 180 94,4
[90,0 ; 97,3]114 3,5
[1,0 ; 8,7]
3, 4 République tchèque 182 84,6
[78,5 ; 89,5]90 2,2
[0,3 ; 7,8]
2, 3 à 4 Amérique latine, 11 pays 393 77,9
[73,8 ; 81,6]341 15,1
[11,7 ; 19,0]
-
Réponse immunitaire chez les nourrissons nés prématurés : - Dans une étude clinique réalisée chez des nourrissons nés prématurés, à 27 semaines de grossesse ou plus, l’immunogénicité de Rotarix a été évaluée dans un sous-groupe de 147 sujets et a montré que Rotarix est immunogène dans cette population ; 85,7 % (IC 95 % : 79,0 ; 90,9) des sujets ont atteint des titres sériques en anticorps IgA anti-rotavirus >= 20 UI/ml (par méthode ELISA), un mois après la seconde dose de vaccin.
PHARMACOCINÉTIQUE
|
L’évaluation des propriétés pharmacocinétiques n’est pas requise pour les vaccins.
SÉCURITE PRÉCLINIQUE
|
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée n’ont pas révélé de risque particulier pour l’homme.
INCOMPATIBILITÉS
|
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
MODALITÉS DE CONSERVATION
|
-
Durée de conservation : - 3 ans.
A conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver dans l’emballage extérieur d’origine, à l’abri de la lumière.
-
Après reconstitution : - Le vaccin doit être administré immédiatement. S’il n’est pas utilisé immédiatement, la conservation en l’état ne doit pas dépasser 24 heures à une température comprise entre 2 °C et 25 °C.
MODALITÉS MANIPULATION/ÉLIMINATION
|
Un dépôt blanc et un surnageant limpide sont observés lors de la conservation de l’applicateur pour administration orale contenant le solvant. Le solvant doit être inspecté visuellement avant et après avoir été agité pour détecter la présence de toute particule étrangère et/ou d’altération de l’aspect physique avant la reconstitution. Le vaccin reconstitué est légèrement plus trouble que le solvant avec un aspect blanc laiteux. Le vaccin reconstitué doit également être inspecté visuellement pour détecter la présence de toute particule étrangère et/ou altération de l’aspect physique avant administration. En cas de non-conformité, jeter le vaccin. Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
-
Instructions pour la reconstitution et l’administration du vaccin : -
-
- Retirer le bouchon plastique du récipient en verre contenant la poudre.
- Connecter l’adaptateur de transfert sur le récipient en verre en le poussant vers le bas jusqu’à ce que l’adaptateur de transfert soit placé correctement et de façon sûre.
- Agiter vigoureusement l’applicateur pour administration orale contenant le solvant. La suspension agitée apparaîtra comme un liquide trouble avec un dépôt blanc sédimentant lentement.
- Retirer le capuchon protecteur de l’applicateur pour administration orale.
- Connecter l’applicateur pour administration orale sur l’adaptateur de transfert en le poussant fermement sur le dispositif.
- Injecter le contenu de l’applicateur pour administration orale dans le récipient en verre contenant la poudre.
- Avec l’applicateur pour administration orale encore attaché, secouer le récipient en verre et l’examiner jusqu’à obtenir une suspension complète de la poudre. Le vaccin reconstitué apparaîtra plus trouble que le solvant seul. Cette apparence est normale.
- Récupérer la totalité du mélange avec l’applicateur pour administration orale.
- Retirer l’applicateur pour administration orale de l’adaptateur de transfert.
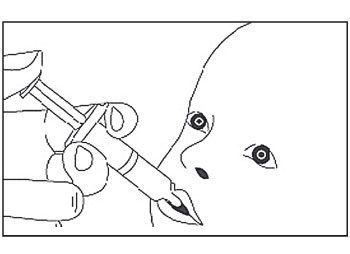
- Le vaccin est destiné à une administration orale uniquement. L’enfant doit être assis en position inclinée. Administrer oralement tout le contenu de l’applicateur pour administration orale (en administrant le contenu complet de l’applicateur pour administration orale à l’intérieur de la joue).
- Ne pas injecter.
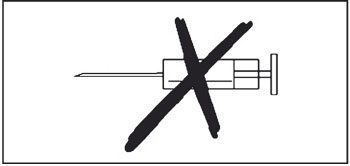
Si le vaccin reconstitué doit être conservé temporairement avant l’administration, replacer le capuchon protecteur sur l’applicateur pour administration orale. L’applicateur pour administration orale contenant le vaccin reconstitué doit être agité à nouveau juste avant l’administration orale. Ne pas injecter.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE
|
LISTE I
| AMM | EU1/05/330/001 ; CIP 3400937232593 (2006, RCP rév 30.03.2010) 1 récipient poudre + 1 applicateur solvant + 1 adaptateur. |
| Non remboursable à la date du 12.10.2010 (demande d’admission à l’étude). |
Titulaire de l’AMM : GlaxoSmithKline Biologicals SA, Rixensart, Belgique.
Laboratoire GlaxoSmithKline
100, route de Versailles. 78163 Marly-le-Roi cdx
Tél : 01 39 17 80 00
Info médic :
Tél : 01 39 17 84 44. Fax : 01 39 17 84 45
Pharmacovigilance : Tél : 01 39 17 80 16


