fulvestrant
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p seringue | |
| Fulvestrant (DCI) | 250 mg |
INDICATIONS |
POSOLOGIE ET MODE D’ADMINISTRATION |
- Femmes adultes (y compris les patientes âgées) :
- La dose recommandée est de 500 mg une fois par mois, avec une dose supplémentaire de 500 mg 2 semaines après la dose initiale.
- Coût du traitement journalier : 30,57 euro(s).
- Population spéciale :
-
- Insuffisance rénale :
- Aucun ajustement posologique n’est recommandé chez les patientes souffrant d’une insuffisance rénale légère à modérée (clairance de la créatinine >= 30 ml/min). La tolérance et l’efficacité n’ont pas été évaluées chez les patientes souffrant d’insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) et, en conséquence, la prudence est recommandée chez ces patientes (cf Mises en garde et Précautions d’emploi).
-
- Insuffisance hépatique :
- Aucun ajustement posologique n’est recommandé chez les patientes atteintes d’une insuffisance hépatique légère à modérée. Cependant, comme l’exposition au fulvestrant peut être augmentée chez ces patientes, Faslodex devra être utilisé avec précaution. Il n’y a pas de données chez les patientes atteintes d’insuffisance hépatique sévère (cf Contre-indications, Mises en garde et Précautions d’emploi, Pharmacocinétique).
- Population pédiatrique :
- La sécurité et l’efficacité de Faslodex n’ont pas été établies chez les enfants âgés de moins de 18 ans. Aucune donnée n’est disponible.
Mode d’administration :
Faslodex doit être administré en 2 injections consécutives de 5 ml par injection intramusculaire lente dans le muscle fessier (1-2 minutes/injection), une dans chaque fesse.
Pour des instructions détaillées sur l’administration, cf Modalités de manipulation et d’élimination.
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active ou à l’un des excipients.
- Grossesse et allaitement (cf Fertilité/Grossesse/Allaitement).
- Insuffisance hépatique sévère (cf Mises en garde et Précautions d’emploi, Pharmacocinétique).
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- Faslodex doit être utilisé avec prudence chez les patientes atteintes d’insuffisance hépatique légère à modérée (cf Posologie et Mode d’administration, Contre-indications, Pharmacocinétique).
- Faslodex doit être utilisé avec prudence chez les patientes souffrant d’insuffisance rénale sévère (clairance de la créatinine inférieure à 30 ml/min).
- En raison de la voie d’administration intramusculaire, Faslodex doit être utilisé avec prudence en cas d’antécédents d’affections hémorragiques, de thrombocytopénie et chez les patientes traitées par des anticoagulants.
- Des événements thromboemboliques sont fréquemment observés chez les patientes atteintes de cancer du sein à un stade avancé et ont été rapportés avec Faslodex dans les études cliniques (cf Effets indésirables). Ceci doit être pris en compte lorsque Faslodex est prescrit à des patientes à risque.
- Il n’y a pas de données sur les effets à long terme du fulvestrant sur les os. Étant donné le mécanisme d’action du fulvestrant, il existe un risque potentiel d’ostéoporose.
INTERACTIONS |
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Les patientes en âge de procréer doivent être informées sur l’utilisation d’une contraception efficace pendant le traitement.
Grossesse :
Faslodex est contre-indiqué lors de la grossesse (cf Contre-indications). Le fulvestrant traverse le placenta après une injection intramusculaire unique chez la rate et la lapine. Des études chez l’animal ont montré une toxicité sur les fonctions de reproduction, y compris une augmentation de l’incidence des anomalies et des morts foetales (cf Sécurité préclinique). En cas de grossesse survenant lors du traitement par Faslodex, la patiente devra être avertie du risque potentiel pour le foetus et du risque potentiel de fausse couche.
Allaitement :
L’allaitement doit être interrompu pendant le traitement par Faslodex. Le fulvestrant est excrété dans le lait de rates qui allaitent. Il n’y a pas de données sur l’excrétion du fulvestrant dans le lait maternel. Compte tenu du risque potentiel d’effets indésirables sévères du fulvestrant pour le nourrisson allaité, l’utilisation en cours d’allaitement est contre-indiquée (cf Contre-indications).
Fécondité :Les effets de Faslodex sur la fécondité dans l’espèce humaine n’ont pas été étudiés.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
| Effets indésirables par système organe classe et fréquence | ||
| Infections et infestations | Fréquent | Infections urinaires |
| Affections du système immunitaire | Fréquent | Réactions d’hypersensibilité |
| Troubles du métabolisme et de la nutrition | Fréquent | Anorexie* |
| Affections du système nerveux | Fréquent | Céphalées |
| Affections vasculaires | Fréquent | Thromboembolies veineuses*, bouffées de chaleur |
| Affections gastro-intestinales | Très fréquent | Nausées |
| Fréquent | Vomissements, diarrhées | |
| Affections hépatobiliaires | Très fréquent | Augmentation des enzymes hépatiques (ALAT, ASAT, phosphatases alcalines)* |
| Affections de la peau et du tissu sous-cutané | Fréquent | Éruptions cutanées |
| Affections musculosquelettiques et systémiques | Fréquent | Douleurs dorsales* |
| Affections des organes de reproduction et du sein | Peu fréquent | Moniliase vaginale, leucorrhée, hémorragie vaginale |
| Troubles généraux et anomalies au site d’administration | Très fréquent | Asthénie*, réactions au site d’injection** |
| Peu fréquent | Hémorragie au site d’injection, hématome au site d’injection | |
* Inclus des effets indésirables pour lesquels l’étendue exacte de la contribution de Faslodex ne peut être évaluée en raison de la maladie sous-jacente.
** Les termes « réactions au site d’injection » n’incluent pas les hémorragies au site d’injection et hématomes au site d’injection.
SURDOSAGE |
Au cours des études chez l’animal, aucun effet autre que ceux liés directement ou indirectement à l’activité anti-estrogène n’a été mis en évidence à des doses plus élevées de fulvestrant (cf Sécurité préclinique).
PHARMACODYNAMIE |
Classe pharmacothérapeutique : Thérapie endocrine, anti-estrogènes (code ATC : L02BA03).
- Mécanisme d’action et effets pharmacodynamiques :
- Le fulvestrant est un antagoniste compétitif des récepteurs aux estrogènes (RE) avec une affinité comparable à l’estradiol. Le fulvestrant bloque les actions trophiques des estrogènes sans posséder une quelconque activité agoniste partielle (de type estrogène).
- Son mécanisme d’action est associé à une diminution des taux d’expression de la protéine du récepteur aux estrogènes
- Des études cliniques menées chez des patientes ménopausées présentant un cancer primaire du sein ont montré que le fulvestrant diminuait significativement l’expression de la protéine RE dans les tumeurs RE positives par comparaison au placebo. Une diminution significative de l’expression des récepteurs à la progestérone a été également observée, en corrélation avec l’absence d’effet estrogénique intrinsèque. Il a été également montré que le fulvestrant 500 mg diminue l’expression de la protéine RE et la prolifération du marqueur Ki67, d’une manière plus importante que le fulvestrant 250 mg en traitement néo-adjuvant des tumeurs mammaires après la ménopause.
- Tolérance et efficacité clinique dans le cancer du sein à un stade avancé :
- Un essai clinique de phase III a été réalisé chez 736 femmes ménopausées atteintes d’un cancer du sein à un stade avancé, dont la maladie a récidivé pendant ou après une hormonothérapie adjuvante ou dont la maladie a progressé suite à une hormonothérapie. L’étude a inclus 423 patientes dont la maladie a récidivé ou progressé sous traitement par anti-estrogène (sous-groupe AE) et 313 patientes dont la maladie a récidivé ou progressé sous traitement par inhibiteur de l’aromatase (sous-groupe IA). Cet essai a comparé l’efficacité et la tolérance de Faslodex 500 mg (n = 362) à Faslodex 250 mg (n = 374). Le critère principal était la survie sans progression (SSP), les critères secondaires clés d’efficacité incluaient le taux de réponse objective (RO%), le bénéfice clinique (BC) et la survie globale (SG). Les résultats d’efficacité de l’étude CONFIRM sont résumés dans le tableau 2.
-
Tableau 2 : Résumé des résultats du critère principal d’efficacité (SSP) et des critères secondaires clés d’efficacité de l’étude CONFIRM Variable Méthode de calcul ; comparaison des traitements Faslodex 500 mg
(N = 362)Faslodex 250 mg
(N = 374)Comparaison entre les groupes (Faslodex 500 mg/ Faslodex 250 mg) Hazard ratio IC 95 % Valeur de p SSP Médiane en mois de K-M ; hazard ratio Population globale 6,5 5,5 0,80 0,68 ; 0,94 0,006 – sous-groupe AE (n = 423)
8,6 5,8 0,76 0,62 ; 0,94 0,013 – sous-groupe IA (n = 313)(a)
5,4 4,1 0,85 0,67 ; 1,08 0,195 SG Médiane en mois de K-M ; hazard ratio Population globale 25,1 22,8 0,84 0,69 ; 1,03 0,091 – sous-groupe AE (n = 423)
27,9 25,9 0,85 0,65 ; 1,13 0,264 – sous-groupe IA (n = 313)(a)
24,1 20,8 0,83 0,62 ; 1,12 0,216 Variable Méthode de calcul ; comparaison des traitements Faslodex 500 mg
(N = 362)Faslodex 250 mg
(N = 374)Comparaison entre les groupes (Faslodex 500 mg/ Faslodex 250 mg) Différence absolue en % IC 95 % RO (%)(b) % de patientes avec une RO ; différence absolue en % Population globale 13,8 14,6 – 0,8 – 5,8 ; 6,3 – sous-groupe AE (n = 296)
18,1 19,1 – 1,0 – 8,2 ; 9,3 – sous-groupe IA (n = 205)(a)
7,3 8,3 – 1,0 – 5,5 ; 9,8 BC (%)(c) % de patientes avec un BC ; différence absolue en % Population globale 45,6 39,6 6,0 – 1,1 ; 13,3 – sous-groupe AE (n = 423)
52,4 45,1 7,3 – 2,2 ; 16,6 – sous-groupe IA (n = 313)(a)
36,2 32,3 3,9 – 6,1 ; 15,2 - SSP : Survie sans progression. RO (%) : Taux de réponse objective. RO : Réponse objective. BC (%) : Pourcentage de bénéfice clinique. BC : Bénéfice clinique. SG : Survie globale. K-M : Kaplan-Meier. IC : Intervalle de confiance. IA : Inhibiteur de l’aromatase. AE : Anti-estrogène.
-
(a)
Faslodex est indiqué chez les patientes dont la maladie a récidivé ou progressé sous traitement par anti-estrogène. Les résultats du sous-groupe IA ne peuvent faire l’objet d’une conclusion.
-
(b)
Le taux de RO a été calculé chez les patientes qui étaient évaluables à l’inclusion (i.e., celles avec une pathologie mesurable à l’inclusion : 240 patientes dans le groupe Faslodex 500 mg et 261 patientes dans le groupe Faslodex 250 mg).
-
(c)
Patientes dont la meilleure réponse objective est soit une réponse complète, soit une réponse partielle, soit une stabilité de la maladie >= 24 semaines.
- Deux essais cliniques de phase III ont été réalisés chez 851 femmes ménopausées atteintes d’un cancer du sein à un stade avancé, dont la maladie a récidivé pendant ou après une hormonothérapie adjuvante ou dont la maladie a progressé suite à une hormonothérapie. 77 % de la population de l’étude présentait un cancer du sein avec des récepteurs aux estrogènes positifs.
- Ces essais ont comparé la tolérance et l’efficacité d’une administration mensuelle de Faslodex 250 mg à celles de l’administration quotidienne de 1 mg d’anastrozole (inhibiteur de l’aromatase).
- Faslodex à la dose mensuelle de 250 mg s’est montré dans son ensemble au moins aussi efficace que l’anastrozole en termes de survie sans progression, de réponse objective, de temps jusqu’au décès. Aucune différence statistiquement significative n’a été observée sur ces critères entre les deux groupes. Le critère principal était la survie sans progression. L’analyse combinée des deux études montre que 83 % des patientes du groupe Faslodex ont vu leur maladie progresser contre 85 % des patientes du groupe anastrozole. L’analyse combinée des deux études montrait que le hazard ratio de Faslodex 250 mg par rapport à l’anastrozole pour la survie sans progression était de 0,95 (IC 95 % 0,82 à 1,10). Le taux de réponse objective était de 19,2 % dans le groupe Faslodex 250 mg, de 16,5 % dans le groupe anastrozole. Le délai médian jusqu’au décès a été de 27,4 mois pour les patientes traitées par Faslodex et de 27,6 mois pour les patientes traitées par l’anastrozole. Le hazard ratio de Faslodex 250 mg par rapport à l’anastrozole pour le temps jusqu’au décès était de 1,01 (IC 95 % 0,86 à 1,19).
- Effets sur l’endomètre postménopausique :
- Les données précliniques ne suggèrent pas un effet stimulant du fulvestrant sur l’endomètre postménopausique (cf Sécurité préclinique). Une étude de 2 semaines réalisée chez des volontaires saines ménopausées traitées par 20 µg/jour d’éthinylestradiol a montré qu’un prétraitement par 250 mg de Faslodex réduit significativement la stimulation de l’endomètre postménopausique comparé à un prétraitement par placebo, évaluée par ultrasons de l’épaisseur de l’endomètre.
- Un traitement néo-adjuvant jusqu’à 16 semaines chez des patientes ayant un cancer du sein traitées par Faslodex 500 mg ou Faslodex 250 mg n’a pas entraîné des modifications cliniquement significatives de l’épaisseur de l’endomètre, indiquant une absence d’effet agoniste. Il n’y a aucune preuve d’effets indésirables sur l’endomètre chez les patientes étudiées atteintes d’un cancer du sein. Aucune donnée n’est disponible sur la morphologie de l’endomètre.
- Dans deux études de court terme (1 et 12 semaines) portant sur des patientes préménopausées souffrant de pathologies gynécologiques bénignes, aucune différence significative au niveau de l’épaisseur de l’endomètre (mesures par ultrasons) n’a été observée entre les groupes fulvestrant et placebo.
- Effets sur l’os :
- Il n’y a pas de données sur les effets à long terme du fulvestrant sur l’os. Un traitement néo-adjuvant jusqu’à 16 semaines chez des patientes ayant un cancer du sein traitées par Faslodex 500 mg ou Faslodex 250 mg n’a pas entraîné des modifications cliniquement significatives au niveau des marqueurs sériques du remodelage osseux.
- Population pédiatrique :
- L’Agence européenne du médicament a différé l’obligation de soumettre les résultats d’études réalisées avec Faslodex dans plusieurs sous-groupes de la population pédiatrique ayant un cancer du sein (cf Posologie et Mode d’administration pour les informations concernant l’usage pédiatrique).
PHARMACOCINÉTIQUE |
- Absorption :
- Après injection intramusculaire de Faslodex à action prolongée, le fulvestrant est absorbé lentement et la concentration plasmatique maximale (Cmax) est atteinte au bout de 5 jours environ. L’administration de Faslodex 500 mg aboutit à des niveaux d’exposition proches de (ou équivalents à) l’état d’équilibre dans le premier mois de l’administration (moyenne [CV] : AUC 475 [33,4 %] ng.jours/ml, Cmax 25,1 [35,1 %] ng/ml, Cmin 16,3 [25,9 %] ng/ml, respectivement). A l’équilibre, les concentrations plasmatiques de fulvestrant se situent dans une fenêtre relativement étroite et présentent jusqu’à environ un facteur 3 entre les concentrations maximales et les concentrations minimales observées. Après injection intramusculaire, les concentrations plasmatiques augmentent proportionnellement à la dose injectée, pour des doses allant de 50 à 500 mg.
- Distribution :
- La distribution du fulvestrant est rapide et importante. Le volume apparent de distribution élevé à l’équilibre (Vdss ) d’environ 3 à 5 l/kg suggère une forte distribution extravasculaire. Le fulvestrant est fortement lié (99 %) aux protéines plasmatiques, principalement aux lipoprotéines de très basse densité (VLDL) de basse densité (LDL) et de haute densité (HDL).
- Aucune étude d’interactions par compétition au niveau des sites de fixation protéique n’a été menée. Le rôle éventuel des globulines se fixant aux hormones sexuelles (SHBG) n’a pas été déterminé.
- Métabolisme :
- Le métabolisme du fulvestrant n’a pas été pleinement étudié mais il implique divers processus éventuels de biotransformation analogues à ceux des stéroïdes endogènes. Les métabolites identifiés (incluant les métabolites 17-cétone, sulfone, 3- sulfate, 3- et 17- glucuronide) présentent une activité inférieure ou similaire à celle du fulvestrant dans les modèles d’études de l’activité anti-estrogénique. Les études réalisées avec des préparations de foie humain et des enzymes humaines recombinantes montrent que parmi les isoenzymes du cytochrome P450, seul le CYP3A4 intervient dans l’oxydation du fulvestrant, alors que des mécanismes autres que la voie P450 semblent prédominants in vivo. Les données obtenues in vitro suggèrent que le fulvestrant n’inhibe pas les isoenzymes du cytochrome P450.
- Élimination :
- Le fulvestrant est principalement éliminé sous forme métabolisée. L’élimination se fait principalement dans les fèces, moins de 1 % de la dose étant éliminé dans les urines. La clairance du fulvestrant est élevée, 11 ml/min/kg ± 1,7, suggérant un taux d’extraction hépatique élevé. La demi-vie terminale (t½) après administration intramusculaire dépend du taux d’absorption et a été estimée à 50 jours.
- Populations spéciales :
- L’analyse de pharmacocinétique de population effectuée sur les données des études de phase III n’a détecté aucune modification du profil pharmacocinétique du fulvestrant en fonction de l’âge (33 à 89 ans), du poids (40 à 127 kg) ou de la race des patientes.
- Insuffisance rénale :
- Chez les patientes présentant une insuffisance rénale légère à modérée, aucune modification cliniquement significative des paramètres pharmacocinétiques du fulvestrant n’a été observée.
- Insuffisance hépatique :
- La pharmacocinétique du fulvestrant a été évaluée en dose unique dans une étude clinique chez des patientes atteintes d’une insuffisance hépatique légère à modérée (scores de Child-Pugh A et B). Une dose élevée d’une formulation injectable intramusculaire, de courte durée d’action, a été utilisée. L’AUC des patientes présentant une insuffisance hépatique a été jusqu’à environ 2,5 fois supérieure à celle des sujets sains.
- Chez les patientes recevant Faslodex, une augmentation de l’exposition de cette amplitude devrait être bien tolérée.
- Les patientes atteintes d’une insuffisance hépatique sévère (score de Child Pugh C) n’ont pas été étudiées.
SÉCURITE PRÉCLINIQUE |
La toxicité aiguë du fulvestrant est faible.
Faslodex, comme les autres formulations de fulvestrant, a été bien toléré dans les espèces animales étudiées lors des études à doses multiples. Des réactions locales au site d’injection, incluant myosite et granulome, ont été attribuées aux excipients, mais la sévérité de la myosite chez le lapin a été plus importante dans le groupe fulvestrant que dans le groupe contrôle (solution saline).
Lors des études de toxicité par administration intramusculaire réitérée menées chez le rat et le chien, la plupart des effets observés, particulièrement les effets sur les organes reproducteurs femelles mais aussi sur les organes hormono-sensibles des deux sexes, ont pu être attribués à l’activité antiestrogénique du fulvestrant. Une artérite au niveau de différents tissus a été observée chez plusieurs chiens après une administration chronique (12 mois).
Dans des études chez des chiens, après administration orale ou intraveineuse, des effets sur le système cardiovasculaire ont été observés : léger allongement du segment S-T de l’électrocardiogramme (voie orale) et pause sinusale chez un chien (voie intraveineuse). Ces effets sont apparus pour des expositions supérieures à celles utilisées chez des patientes (Cmax > 15 fois) et sont considérés comme peu significatifs en matière de sécurité d’emploi pour l’espèce humaine aux doses cliniques.
Le fulvestrant n’a montré aucun potentiel génotoxique.
Les effets constatés, à des doses similaires aux doses cliniques, sur la reproduction et sur le développement embryonnaire et foetal sont la conséquence de l’activité antiestrogène du fulvestrant.
Chez les rats, une diminution réversible de la fertilité des femelles, une diminution de la survie embryonnaire, une dystocie et une augmentation de la fréquence des anomalies foetales y compris de la courbure du tarse ont été observées. Chez des lapines ayant reçu du fulvestrant, la gestation n’a pu être maintenue. Une augmentation du poids du placenta et des pertes post-implantatoires ont été observés. Chez les lapines, il y a eu une augmentation de l’incidence des modifications foetales (bascule en arrière du bassin et de la vertèbre présacrée 27).
Une étude de cancérogénicité de deux ans chez le rat (administration intramusculaire de Faslodex) a mis en évidence une augmentation de la fréquence des tumeurs ovariennes bénignes des cellules de la granulosa chez les femelles pour des doses de 10 mg/rat/15 jours, et une augmentation des tumeurs des cellules testiculaires de Leydig chez les mâles.
L’induction de telles tumeurs est en rapport avec les effets pharmacologiques endocriniens. Ces effets sont dépourvus de signification clinique dans le cadre de l’utilisation du fulvestrant chez des femmes ménopausées souffrant d’un cancer du sein au stade avancé.
INCOMPATIBILITÉS |
Vu l’absence d’études de compatibilité, ce médicament ne doit être mélangé avec aucun autre médicament.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 4 ans.
A conserver au réfrigérateur (entre 2 °C et 8 °C).
Conserver la seringue préremplie dans l’emballage d’origine pour protéger de la lumière.
MODALITÉS MANIPULATION/ÉLIMINATION |
- Instructions pour l’administration :
- Avertissement : Ne jamais stériliser à l’autoclave l’aiguille protégée (aiguille protégée hypodermique BD SafetyGlide) avant utilisation. Pendant toute la durée de l’utilisation et de la procédure d’élimination de l’aiguille, les mains doivent toujours rester derrière l’aiguille.
- Pour chacune des deux seringues :
- Retirer le corps de la seringue en verre du plateau-support et vérifier qu’il n’a pas été endommagé.
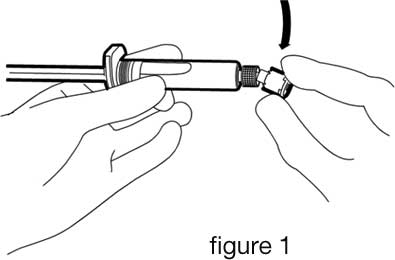
- Rompre l’opercule de la protection en plastique blanc de la connexion Luer de la seringue pour enlever le protège-embout en caoutchouc qui est attaché à son extrémité (cf figure 1).
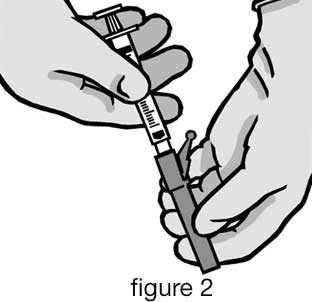
- Retirer l’emballage extérieur de l’aiguille protégée (BD SafetyGlide). Fixer l’aiguille protégée sur la connexion Luer (cf figure 2).
- Tourner fermement jusqu’à fixation.
- Fixer par rotation l’aiguille sur la connexion Luer.
- Débloquer le protège-aiguille en tirant d’un coup sec afin de ne pas en endommager la pointe de l’aiguille.
- Ne retirer le protège-aiguille qu’au-dessus du site d’injection.
- Les solutions à usage parentéral doivent être contrôlées visuellement avant administration afin de détecter la présence de particules ou un changement de coloration.
- Chasser les bulles de la seringue.
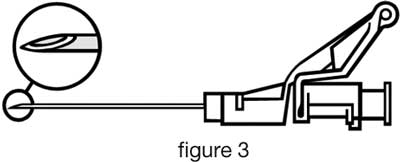
- Injecter lentement en intramusculaire (1-2 minutes/injection) dans le muscle fessier. Pour plus de facilité, le biseau de l’aiguille est orienté du côté du bras du levier (cf figure 3).
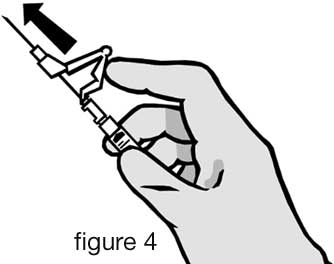
- Après injection, enclencher immédiatement le dispositif de protection de l’aiguille en poussant à fond le bras de levier avec le doigt (cf figure 4).
- Note : Tenir la seringue écartée de soi-même et d’autrui pour enclencher la protection. Écouter le clic et vérifier visuellement que l’aiguille a bien été complètement recouverte.
- Retirer le corps de la seringue en verre du plateau-support et vérifier qu’il n’a pas été endommagé.
 |
 |
 |
 |
- Élimination :
- Les seringues préremplies sont exclusivement à usage unique.
- Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM | EU/1/03/269/001 ; CIP 3400936349001 (2004, RCP rév 25.10.2010). |
| Prix : | 458.56 euros (1 seringue). |
| Remb Séc soc à 100 %. Collect. | |
| Prix ou tarif de responsabilité (HT) par UCD : | UCD 9261073 (seringue) : 400.00 euros. |
| Inscrit sur la liste des spécialités prises en charge en sus des GHS. | |
Titulaire de l’AMM : AstraZeneca UK Limited, Alderley Park, Macclesfield, Cheshire, SK10 4TG, Royaume-Uni.
AstraZeneca
1, place Renault. 92844 Rueil-Malmaison cdx
Tél : 01 41 29 40 00. Fax : 01 41 29 40 01
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


