ranibizumab
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p flacon | |
| Ranibizumab* (DCI) | 2,3 mg |
INDICATIONS |
- le traitement de la forme néovasculaire (humide) de la dégénérescence maculaire liée à l’âge (DMLA), cf Pharmacodynamie ;
- le traitement de la baisse visuelle due à l’oedème maculaire diabétique (OMD), cf Pharmacodynamie.
POSOLOGIE ET MODE D’ADMINISTRATION |
- Traitement de la DMLA néovasculaire :
- Dans la DMLA néovasculaire, la dose recommandée de Lucentis est de 0,5 mg, administrée une fois par mois en une injection intravitréenne unique. Cette dose correspond à un volume d’injection de 0,05 ml.
- Le traitement par Lucentis commence par une phase d’induction avec 1 injection par mois pendant 3 mois consécutifs, suivie d’une phase de maintien au cours de laquelle l’acuité visuelle des patients sera contrôlée une fois par mois. Si le patient présente une perte d’acuité visuelle de plus de 5 lettres (échelle ETDRS ou équivalent d’une ligne sur l’échelle de Snellen), Lucentis peut être administré. L’intervalle entre deux doses ne doit pas être inférieur à 1 mois.
- Traitement de la baisse visuelle due à l’OMD :
- En cas de baisse visuelle due à un OMD, la dose recommandée de Lucentis est de 0,5 mg, administrée en une injection intravitréenne unique. Cette dose correspond à un volume d’injection de 0,05 ml.
- Le traitement sera administré une fois par mois et poursuivi jusqu’à ce que l’acuité visuelle maximale soit atteinte, c’est-à-dire jusqu’à ce que l’acuité visuelle du patient soit stable lors de trois évaluations mensuelles consécutives effectuées au cours du traitement par le ranibizumab. Si aucune amélioration de l’acuité visuelle n’est constatée à l’issue d’une série de trois injections, la poursuite du traitement n’est pas recommandée.
- Par la suite, l’acuité visuelle doit être contrôlée une fois par mois.
- En cas de nouvelle baisse de l’acuité visuelle due à l’OMD constatée lors d’un contrôle, le traitement doit être réinstauré. Des injections mensuelles doivent alors être réalisées jusqu’à ce que l’acuité visuelle soit à nouveau stable lors de trois évaluations mensuelles consécutives (ceci impliquant un minimum de deux injections). L’intervalle entre deux doses ne doit pas être inférieur à 1 mois.
- Lucentis et photocoagulation au laser dans l’OMD :
- Des données concernant l’administration concomitante de Lucentis et d’une photocoagulation au laser sont disponibles (cf Pharmacodynamie). Si les deux traitements sont réalisés le même jour, Lucentis doit être administré au moins 30 minutes après la photocoagulation au laser. Lucentis peut être administré aux patients ayant été traités précédemment par photocoagulation au laser.
- Groupes de patients particuliers :
-
- Insuffisance hépatique :
- Lucentis n’a pas été étudié chez les patients présentant une insuffisance hépatique. Cependant, aucune précaution particulière n’est nécessaire pour cette population.
-
- Insuffisance rénale :
- Aucune adaptation de la dose n’est nécessaire chez les patients présentant une insuffisance rénale (cf Pharmacocinétique).
-
- Population pédiatrique :
- Lucentis ne doit pas être utilisé chez l’enfant et l’adolescent suite à un manque de données concernant la sécurité et l’efficacité dans ces sous-groupes de patients.
-
- Patients âgés :
- Aucune adaptation de la dose n’est nécessaire chez les patients âgés. L’expérience chez les patients âgés de plus de 75 ans présentant un OMD est limitée.
-
- Origine ethnique :
- L’expérience avec ce traitement est limitée chez les personnes autres que celles d’origine caucasienne.
Mode d’administration :
Comme pour tous les médicaments à usage parentéral, Lucentis doit être contrôlé visuellement avant l’administration pour vérifier l’absence de particules et de changement de coloration.
Avant le traitement, le patient doit être informé qu’il doit s’autoadministrer un collyre antibactérien (4 fois par jour pendant 3 jours avant et après chaque injection).
La procédure d’injection doit être réalisée en conditions d’asepsie, incluant la désinfection chirurgicale des mains, le port de gants stériles, l’utilisation d’un champ stérile et d’un spéculum à paupières stérile (ou équivalent) et la possibilité d’effectuer une paracentèse stérile (si nécessaire). Les antécédents médicaux du patient relatifs aux réactions d’hypersensibilité doivent être attentivement évalués avant de procéder à l’administration intravitréenne (cf Mises en garde et Précautions d’emploi). La peau autour de l’oeil, la paupière et la surface oculaire doivent être désinfectées et une anesthésie appropriée et un antibactérien local à large spectre doivent être administrés avant l’injection.
Pour toute information concernant la préparation de Lucentis, cf Modalités de manipulation et d’élimination.
L’aiguille pour injection doit être introduite 3,5-4,0 mm en arrière du limbe dans la cavité vitréenne, en évitant le méridien horizontal et en visant le milieu du globe oculaire. Le volume de 0,05 ml peut alors être injecté ; un point d’injection scléral différent doit être utilisé lors des injections ultérieures.
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active ou à l’un des excipients.
- Patients présentant une infection oculaire ou périoculaire active ou suspectée.
- Patients présentant une inflammation intraoculaire active sévère.
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- diminution de la meilleure acuité visuelle corrigée (MAVC) d’au moins 30 lettres par rapport à la dernière évaluation de l’acuité visuelle ;
- pression intraoculaire >= 30 mm Hg ;
- déchirure rétinienne ;
- hémorragie sous-rétinienne impliquant le centre de la fovéa ou lorsque la taille de l’hémorragie est supérieure ou égale à 50 % de la surface totale de la lésion ;
- chirurgie intraoculaire effectuée au cours des 28 jours précédents ou prévue au cours des 28 jours à venir.
INTERACTIONS |
Aucune étude spécifique d’interaction n’a été réalisée.
Pour le traitement concomitant par photocoagulation au laser et Lucentis dans l’OMD, cf Posologie et Mode d’administration, Pharmacodynamie.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement.
Grossesse :
Il n’existe pas de données cliniques sur l’utilisation du ranibizumab chez la femme enceinte. Les études chez le singe cynomolgus n’ont pas montré d’effets délétères directs ou indirects sur la gestation ou le développement embryonnaire ou foetal (cf Sécurité préclinique). L’exposition systémique au ranibizumab est attendue comme très faible après une administration oculaire, mais, compte tenu de son mécanisme d’action, le ranibizumab doit être considéré comme potentiellement tératogène et embryo/foetotoxique. Par conséquent, le ranibizumab ne doit pas être utilisé pendant la grossesse à moins que le bénéfice prévisible pour la mère ne l’emporte sur le risque potentiel pour le foetus. Chez les femmes traitées par le ranibizumab qui envisagent une grossesse, il est recommandé d’attendre au moins 3 mois après la dernière administration de ranibizumab.
Allaitement :
On ne sait pas si Lucentis est excrété dans le lait maternel. L’allaitement n’est pas recommandé durant l’utilisation de Lucentis.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
- Population présentant une DMLA néovasculaire :
- Dans la DMLA, au total, 1315 patients ont constitué la population d’évaluation de la tolérance dans les trois études de phase III avec 24 mois d’exposition à Lucentis et 440 patients ont été traités à la dose recommandée de 0,5 mg.
- Les événements indésirables graves liés à la procédure d’injection comprennent des endophtalmies, des décollements rhegmatogènes de la rétine, des déchirures rétiniennes et des cataractes traumatiques iatrogènes (cf Mises en garde et Précautions d’emploi).
- Les autres événements oculaires graves observés chez les patients traités par Lucentis comprennent des inflammations intraoculaires et des élévations de la pression intraoculaire (cf Mises en garde et Précautions d’emploi).
- Les événements indésirables listés ci-dessous sont survenus à une incidence plus élevée (d’au moins 2 %) chez les patients traités par Lucentis 0,5 mg comparativement à ceux recevant le traitement contrôle (injection simulée ou PDT par la vertéporfine) dans les trois études contrôlées de phase III FVF2598g (Marina), FVF2587g (Anchor) et FVF3192g (Pier) dans la DMLA néovasculaire. Ils ont donc été considérés comme des effets indésirables potentiels du médicament. Les données de tolérance décrites ci-dessous incluent également tous les événements indésirables (chez au moins 0,5 % des patients) suspectés d’être au moins potentiellement liés à la procédure d’injection ou au médicament chez les 440 patients atteints de DMLA néovasculaire (des 3 études combinées) traités par 0,5 mg.
- Les événements indésirables sont listés par classe de systèmes d’organes et fréquence en utilisant la convention suivante : très fréquent (>= 1/10), fréquent (>= 1/100, < 1/10), peu fréquent (>= 1/1000, < 1/100), rare (>= 1/10 000, < 1/1000) et très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque fréquence de groupe, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité.
- Population présentant un OMD :
- La tolérance de Lucentis a été étudiée dans une étude contrôlée contre injections simulées d’une durée d’un an (Resolve) et dans une étude contrôlée contre photocoagulation au laser d’une durée d’un an (Restore), conduites respectivement chez 102 et 235 patients présentant une baisse visuelle due à un OMD et traités par le ranibizumab (cf Pharmacodynamie). Seul l’événement « infections des voies urinaires » a été classé dans la catégorie « Fréquent » de la liste d’effets indésirables ci-dessous, alors que la fréquence et la sévérité des autres événements oculaires et non oculaires rapportés au cours des études Resolve et Restore ont été similaires à celles observées dans les études réalisées dans la DMLA néovasculaire.
- Très fréquent : rhinopharyngite.
- Fréquent : infections des voies urinaires*.
- Fréquent : anémie.
- Fréquent : hypersensibilité.
- Fréquent : anxiété.
- Très fréquent : céphalées.
- Très fréquent : hyalite, décollement du vitré, hémorragie rétinienne, trouble visuel, douleur oculaire, corps flottants vitréens, hémorragie conjonctivale, irritation oculaire, sensation de corps étranger dans l’oeil, sécrétion lacrymale accrue, blépharite, sécheresse oculaire, hyperhémie oculaire, prurit oculaire.
- Fréquent : dégénérescence rétinienne, affection de la rétine, décollement de la rétine, déchirure rétinienne, décollement de l’épithélium pigmentaire rétinien, déchirure de l’épithélium pigmentaire rétinien, baisse de l’acuité visuelle, hémorragie vitréenne, affection vitréenne, uvéite, iritis, iridocyclite, cataracte, cataracte sous-capsulaire, opacification de la capsule postérieure, kératite ponctuée, abrasion de la cornée, effet Tyndall dans la chambre antérieure, vision trouble, hémorragie au point d’injection, hémorragie oculaire, conjonctivite, conjonctivite allergique, sécrétions oculaires, photopsie, photophobie, gêne oculaire, oedème palpébral, douleur palpébrale, hyperhémie conjonctivale.
- Peu fréquent : cécité, endophtalmie, hypopyon, hyphéma, kératopathie, synéchie de l’iris, dépôts cornéens, oedème cornéen, stries cornéennes, douleur au point d’injection, irritation au point d’injection, sensation intraoculaire anormale, irritation palpébrale.
- Fréquent : toux.
- Fréquent : nausées.
- Fréquent : réactions cutanées de type allergique (rash, urticaire, prurit, érythème).
- Très fréquent : arthralgie.
- Très fréquent : augmentation de la pression intraoculaire.
* Observé uniquement dans la population OMD.
- Effets indésirables liés à la classe :
- Au cours des études de phase III dans la DMLA néovasculaire, la fréquence globale des hémorragies non oculaires, un effet indésirable potentiellement lié à l’inhibition systémique du VEGF (facteur de croissance de l’endothélium vasculaire), était légèrement augmentée chez les patients traités par le ranibizumab. Cependant, il n’existait aucune homogénéité parmi les différentes hémorragies. Il existe un risque théorique d’événements thromboemboliques artériels suite à l’utilisation intravitréenne des inhibiteurs du VEGF. Un taux d’incidence faible d’événements thromboemboliques artériels a été observé dans les essais cliniques menés avec Lucentis chez les patients atteints de DMLA et d’OMD, et aucune différence majeure n’a été constatée entre les groupes traités par le ranibizumab comparativement aux groupes contrôles.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : médicament contre la néovascularisation (code ATC : S01LA04).
Le ranibizumab est un fragment d’anticorps monoclonal humanisé recombinant dirigé contre le facteur de croissance de l’endothélium vasculaire humain de type A (VEGF-A). Il se lie avec une haute affinité aux isoformes du VEGF-A (par exemple VEGF110, VEGF121 et VEGF165), empêchant dès lors la liaison du VEGF-A à ses récepteurs VEGFR-1 et VEGFR-2. La liaison du VEGF-A à ses récepteurs induit une prolifération des cellules endothéliales et une néovascularisation ainsi qu’une perméabilité vasculaire, tous ces facteurs étant considérés comme contribuant à la progression de la forme néovasculaire de la dégénérescence maculaire liée à l’âge et de l’oedème maculaire diabétique responsable d’une altération visuelle.
- Traitement de la DMLA néovasculaire :
- Dans la DMLA néovasculaire, la tolérance et l’efficacité cliniques de Lucentis ont été évaluées dans trois études randomisées d’une durée de 24 mois, en double insu, contrôlées, comparativement à une injection simulée ou un traitement actif chez des patients atteints de DMLA néovasculaire. Au total, 1323 patients (879 traités par un traitement actif et 444 par injection simulée) ont été inclus dans ces études.
- Dans l’étude FVF2598g (Marina), 716 patients atteints de DMLA au stade de néovascularisation choroïdienne (NVC) visible minoritaire (« minimally classic ») ou occulte pure ont reçu des injections intravitréennes mensuelles de Lucentis 0,3 mg (n = 238) ou 0,5 mg (n = 240) ou des injections simulées (n = 238).
- Dans l’étude FVF2587g (Anchor), 423 patients atteints de DMLA au stade de néovascularisation choroïdienne (NVC) à prédominance visible ont reçu soit 1) des injections intravitréennes mensuelles de Lucentis 0,3 mg et une PDT simulée (n = 140), 2) des injections intravitréennes mensuelles de Lucentis 0,5 mg et une PDT simulée (n = 140) ou 3) des injections intravitréennes simulées et une PDT active par la vertéporfine (n = 143). La PDT, simulée ou active par la vertéporfine, a été administrée avec l’injection initiale de Lucentis puis tous les 3 mois si l’angiographie à la fluorescéine montrait la persistance ou la réapparition d’une diffusion vasculaire.
- Dans les deux études, le critère principal d’évaluation de l’efficacité était la proportion de patients ayant conservé leur vision, définis comme les patients ayant perdu < 15 lettres d’acuité visuelle à 12 mois par rapport à l’acuité visuelle initiale. Environ 95 % des patients traités par Lucentis ont conservé leur acuité visuelle. 34 à 40 % des patients traités par Lucentis ont présenté une amélioration cliniquement significative de la vision, définie comme un gain d’au moins 15 lettres à 12 mois (cf tableaux 1 et 2).
-
Tableau 1 : Résultats à 12 mois et à 24 mois dans l’étude FVF2598g (Marina) Mesure du résultat Mois Injection simulée
(n = 238)Lucentis 0,5 mg
(n = 240)Perte < 15 lettres d’acuité visuelle (%) (a)
(conservation de la vision)Mois 12 62 % 95 % Mois 24 53 % 90 % Gain >= 15 lettres d’acuité visuelle (%) (a) Mois 12 5 % 34 % Mois 24 4 % 33 % Variation moyenne de l’acuité visuelle [lettres] (ET) (a) Mois 12 – 10,5 (16,6) + 7,2 (14,4) Mois 24 – 14,9 (18,7) + 6,6 (16,5) -
(a)
p < 0,01
-
Tableau 2 : Résultats à 12 mois et à 24 mois dans l’étude FVF2587g (Anchor) Mesure du résultat Mois PDT par la vertéporfine
(n = 143)Lucentis 0,5 mg
(n = 140)Perte < 15 lettres d’acuité visuelle (%) (a)
(conservation de la vision)Mois 12 64 % 96 % Mois 24 66 % 90 % Gain >= 15 lettres d’acuité visuelle (%) (a) Mois 12 6 % 40 % Mois 24 6 % 41 % Variation moyenne de l’acuité visuelle [lettres] (ET) (a) Mois 12 – 9,5 (16,4) + 11,3 (14,6) Mois 24 – 9,8 (17,6) + 10,7 (16,5) -
(a)
p < 0,01
-
Figure 1 : Variation moyenne de l’acuité visuelle à 24 mois dans l’étude FVF2598g (Marina) et dans l’étude FVF2587g (Anchor), par rapport à l’acuité visuelle initiale 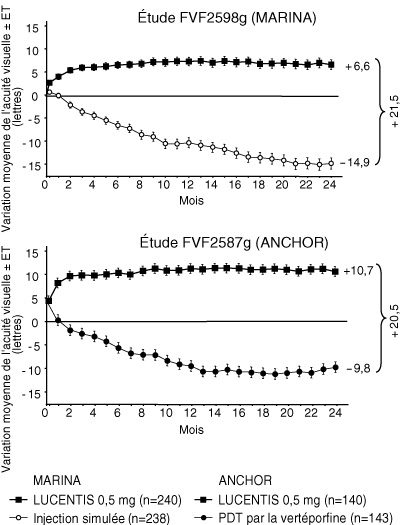
- Les résultats des deux études montrent que la poursuite du traitement par le ranibizumab peut également présenter un bénéfice chez les patients ayant perdu >= 15 lettres de meilleure acuité visuelle corrigée (MAVC) au cours de la première année de traitement.
- L’utilisation de Lucentis au-delà de 24 mois n’a pas été étudiée.
- L’étude FVF3192g (Pier) est une étude randomisée, en double insu, contrôlée contre des injections simulées, destinée à évaluer la tolérance et l’efficacité de Lucentis chez 184 patients présentant toutes les formes de DMLA néovasculaire. Les patients ont reçu des injections intravitréennes de Lucentis 0,3 mg (n = 60) ou 0,5 mg (n = 61) ou des injections simulées (n = 63) une fois par mois, à raison de 3 doses consécutives, suivies d’une dose administrée une fois tous les 3 mois. A partir du mois 14 de l’étude, les patients recevant des injections simulées avaient la possibilité de recevoir Lucentis, et à partir du mois 19, la fréquence des traitements pouvait être augmentée. Les patients traités par Lucentis dans l’étude Pier ont reçu en moyenne 10 traitements.
- Le critère principal d’évaluation de l’efficacité était la variation moyenne de l’acuité visuelle à 12 mois par rapport à l’acuité visuelle initiale. Après une augmentation initiale (suivant l’administration de doses mensuelles), l’acuité visuelle des patients a diminué en moyenne avec des administrations trimestrielles, pour revenir à la valeur initiale au mois 12 et cet effet a été conservé à 24 mois chez la plupart des patients traités par le ranibizumab (82 %). Des données recueillies chez un nombre limité de patients ayant été traités par Lucentis après avoir reçu des injections simulées pendant plus d’un an suggèrent qu’une initiation précoce du traitement serait associée à une meilleure préservation de l’acuité visuelle.
- Les données d’une étude en ouvert (Protect), au cours de laquelle la tolérance d’une PDT par la vertéporfine et d’une administration de Lucentis 0,5 mg réalisée le même jour a été évaluée chez 32 patients suivis pendant 9 mois, ont montré une incidence de l’inflammation intraoculaire après traitement initial de 6,3 % (2 patients sur 32).
- Dans les deux études Marina et Anchor, l’amélioration de l’acuité visuelle observée avec Lucentis 0,5 mg à 12 mois a été accompagnée de bénéfices rapportés par le patient. Ils ont été estimés grâce au questionnaire de la fonction visuelle (VFQ-25) du National Eye Institute, qui étaient des critères secondaires d’évaluation d’efficacité préspécifiés. Les différences entre les groupes Lucentis 0,5 mg et les deux groupes contrôles ont été estimées avec des valeurs de p comprises entre 0,009 et < 0,0001.
- Traitement de la baisse visuelle due à l’OMD :
- L’efficacité et la tolérance de Lucentis ont été évaluées au cours de deux études d’une durée de 12 mois, randomisées, en double insu, contrôlées comparativement à une injection simulée ou un traitement actif, conduites chez des patients présentant une baisse visuelle due à un oedème maculaire diabétique. Au total, 496 patients (336 patients recevant le traitement actif et 160 patients dans les groupes témoins) ont été inclus dans ces études. La majorité des patients inclus étaient des patients diabétiques de type 2 ; 28 patients traités par le ranibizumab étaient des patients diabétiques de type 1.
- Dans l’étude de phase II D2201 (Resolve), 151 patients ont reçu des injections intravitréennes mensuelles de ranibizumab (6 mg/ml, n = 51 ; 10 mg/ml, n = 51) ou des injections simulées (n = 49) jusqu’à ce que les critères prédéfinis d’arrêt du traitement soient atteints. La dose initiale de ranibizumab (0,3 mg ou 0,5 mg) pouvait être doublée à tout moment au cours de l’étude après la première injection. Pendant l’étude, la photocoagulation au laser était autorisée comme traitement de secours à tout moment après le troisième mois dans les deux groupes de traitement. L’étude comportait deux parties : une partie exploratoire (les 42 premiers patients évalués à 6 mois) et une partie de confirmation (les 109 autres patients évalués au douzième mois).
- Dans la partie de confirmation de l’étude (2/3 des patients), le critère d’efficacité principal, qui était la variation moyenne de la MAVC du mois 1 au mois 12 par rapport aux valeurs initiales, a été statistiquement supérieur chez les patients traités par le ranibizumab par rapport à ceux traités par injections simulées, un résultat également confirmé dans la population totale de l’étude, avec une moyenne de 10 injections. La supériorité de l’effet du traitement a également été démontrée pour les principaux critères secondaires relatifs à la MAVC : variation moyenne de la MAVC à 12 mois et pourcentage de patients présentant un gain de MAVC >= 10 lettres et >= 15 lettres à 12 mois (cf tableau 3).
-
Tableau 3 : Résultats à 12 mois dans l’étude D2201 (Resolve) – Population totale de l’étude Résultat Groupes ranibizumab combinés
(n = 102)Injections simulées
(n = 49)Variation moyenne de la MAVC du mois 1 au mois 12 par rapport aux valeurs initiales (lettres) [ET] +7,8 (7,72) -0,1 (9,77) Valeur de p < 0,0001 Variation moyenne de la MAVC au mois 12 (lettres) [ET] +10,3 (9,14) -1,4 (14,16) Valeur de p < 0,0001 Gain >= 10 lettres de MAVC (% de patients) au mois 12 60,8 18,4 Valeur de p < 0,0001 Gain >= 15 lettres de MAVC (% de patients) au mois 12 32,4 10,2 Valeur de p 0,0043 - Dans l’étude de phase III D2301 (Restore), 345 patients présentant une baisse visuelle due à un oedème maculaire ont été randomisés et ont reçu soit une injection intravitréenne de ranibizumab 0,5 mg en monothérapie et une photocoagulation au laser simulée (n = 116), soit un traitement combiné par ranibizumab 0,5 mg et une photocoagulation au laser (n = 118), ou une injection simulée et une photocoagulation au laser (n = 111). Le traitement par le ranibizumab a débuté avec des injections intravitréennes mensuelles et a été poursuivi jusqu’à ce que l’acuité visuelle soit stable au moins lors de trois évaluations mensuelles consécutives. Le traitement était réinstauré lorsqu’une diminution de la MAVC due à l’aggravation de l’OMD était observée. La photocoagulation au laser a été administrée au début de l’étude, le même jour et au moins 30 minutes avant l’injection de ranibizumab, puis selon les besoins conformément aux critères ETDRS.
- Le critère principal d’efficacité qui était la variation moyenne de la MAVC du mois 1 au mois 12 par rapport aux valeurs initiales a été supérieur chez les patients recevant le ranibizumab en monothérapie ou en traitement adjuvant de la photocoagulation au laser comparativement au groupe témoin photocoagulation au laser seule, sans différences significatives en termes d’efficacité entre le ranibizumab en monothérapie et en traitement adjuvant de la photocoagulation au laser, avec une moyenne de 7 injections. Une amélioration cliniquement significative de la MAVC – gain d’au moins 10 lettres et d’au moins 15 lettres – a également été observée, ainsi qu’une supériorité de la variation moyenne de la MAVC à 12 mois (cf tableau 4 et figure 2).
-
Tableau 4 : Résultats à 12 mois dans l’étude D2301 (Restore) Résultat exprimé par rapport aux valeurs initiales Ranibizumab 0,5 mg
(n = 115)Ranibizumab 0,5 mg + photocoagulation au laser
(n = 118)Photocoagulation au laser
(n = 110)Variation moyenne de la MAVC du mois 1 au mois 12 (± ET) 6,1 (6,4) 5,9 (7,9) 0,8 (8,6) Valeur de p < 0,0001 < 0,0001 Gain >= 10 lettres ou MAVC >= 84 (% de patients) 37,4 43,2 15,5 Valeur de p < 0,0001 < 0,0001 Gain >= 15 lettres ou MAVC >= 84 (% de patients) 22,6 22,9 8,2 Valeur de p 0,0032 0,0021 -
Figure 2 : Variation moyenne de l’acuité visuelle au cours du temps dans l’étude D2301 (Restore) 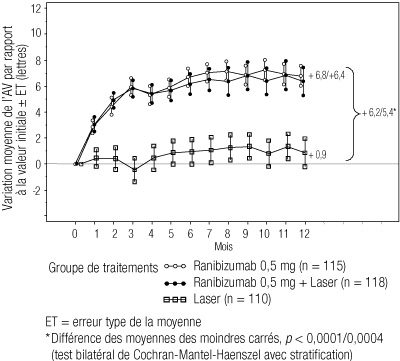
- Cet effet a été retrouvé dans la plupart des sous-groupes. Cependant, les patients présentant une MAVC relativement bonne au début de l’étude (> 73 lettres) associée à un oedème maculaire avec une épaisseur centrale de la rétine inférieure à 300 µm n’ont pas semblé tirer de bénéfice du traitement par le ranibizumab comparativement à la photocoagulation au laser.
- L’amélioration de l’acuité visuelle observée avec Lucentis 0,5 mg à 12 mois s’est traduite par des bénéfices rapportés par le patient au regard de la plupart des fonctions liées à la vision incluant le score composite global et plus particulièrement les critères de vision générale et d’activités liées à la vision de près et à la vision de loin, mesurés par les scores du questionnaire de la fonction visuelle (VFQ-25) du National Eye Institute, qui étaient un critère secondaire d’évaluation de l’efficacité préspécifié. Aucune différence entre les traitements n’a pu être établie pour les autres domaines de ce questionnaire. La différence entre Lucentis 0,5 mg et le groupe témoin a été objectivée par des valeurs de p de 0,0137 (ranibizumab monothérapie) et 0,0041 (ranibizumab + laser) pour le score composite VFQ-25.
- Dans les deux études, l’amélioration de la vision a été accompagnée d’une réduction continue de l’oedème maculaire appréciée par la mesure de l’épaisseur centrale de la rétine (ECR).
- Aucun nouvel effet indésirable oculaire ou non oculaire n’a été observé dans aucune des études décrites ci-dessus menées chez des patients atteints d’OMD.
- Population pédiatrique :
- La sécurité d’emploi et l’efficacité du ranibizumab n’ont pas encore été étudiées dans ce groupe de patients.
- L’Agence européenne du médicament a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec Lucentis dans tous les sous-groupes de la population pédiatrique pour le traitement de la baisse visuelle due à l’oedème maculaire diabétique (cf Posologie et Mode d’administration pour les informations concernant l’usage pédiatrique).
PHARMACOCINÉTIQUE |
Après une administration intravitréenne mensuelle de Lucentis à des patients atteints de DMLA néovasculaire, les concentrations sériques de ranibizumab ont été généralement faibles, les concentrations maximales (Cmax) étant généralement inférieures à la concentration de ranibizumab nécessaire pour inhiber de 50 % l’activité biologique du VEGF (11-27 ng/ml, évaluée par un essai de prolifération cellulaire in vitro). La Cmax a été proportionnelle à la dose sur l’intervalle de doses allant de 0,05 à 1,0 mg/oeil. Les concentrations sériques mesurées chez un nombre limité de patients atteints d’OMD montrent qu’une exposition systémique légèrement supérieure à celle observée chez les patients atteints de DMLA néovasculaire ne peut être exclue.
Sur la base d’une analyse pharmacocinétique de population et de la disparition du ranibizumab du sérum chez les patients atteints de DMLA néovasculaire traités à la dose de 0,5 mg, la demi-vie d’élimination vitréenne moyenne du ranibizumab est d’environ 9 jours. Après une administration intravitréenne mensuelle de Lucentis 0,5 mg/oeil, la Cmax sérique du ranibizumab, atteinte environ 1 jour après l’administration, devrait généralement être comprise entre 0,79 et 2,90 ng/ml et la Cmin comprise entre 0,07 et 0,49 ng/ml. Les concentrations sériques de ranibizumab devraient être environ 90 000 fois plus faibles que les concentrations vitréennes de ranibizumab.
- Patients insuffisants rénaux :
- Aucune étude spécifique n’a été conduite pour évaluer la pharmacocinétique de Lucentis chez les patients présentant une insuffisance rénale. Lors d’une analyse pharmacocinétique de population, 68 % des patients (136/200) présentaient une insuffisance rénale (46,5 % légère [50-80 ml/min], 20 % modérée [30-50 ml/min] et 1,5 % sévère [< 30 ml/min]). La clairance systémique a été légèrement plus faible, mais cette différence n’a pas été cliniquement significative.
- Insuffisance hépatique :
- Aucune étude spécifique n’a été conduite pour évaluer la pharmacocinétique de Lucentis chez les patients présentant une insuffisance hépatique.
SÉCURITE PRÉCLINIQUE |
Après administration intravitréenne bilatérale de ranibizumab à des singes Cynomolgus à des doses comprises entre 0,25 mg/oeil et 2,0 mg/oeil, une fois toutes les 2 semaines pendant 26 semaines, des effets oculaires dose-dépendants ont été observés.
Au niveau intraoculaire, des augmentations dose-dépendantes de l’effet Tyndall protéique et cellulaire ont été observées dans la chambre antérieure, avec un pic deux jours après l’injection. La sévérité de la réponse inflammatoire a généralement diminué lors des injections ultérieures ou pendant la période de récupération. Dans le segment postérieur, une infiltration cellulaire et des corps flottants ont été observés dans le vitré, qui ont également eu tendance à être dose-dépendants et qui ont généralement persisté jusqu’à la fin de la période de traitement. Dans l’étude de 26 semaines, la sévérité de l’inflammation vitréenne a augmenté avec le nombre d’injections. Toutefois, des signes de réversibilité ont été observés après la période de récupération. La nature et la chronologie de l’inflammation du segment postérieur sont évocatrices d’une réponse anticorps à médiation immunitaire, qui peut être cliniquement non pertinente. La formation de cataractes a été observée chez certains animaux après une période relativement longue d’inflammation intense, suggérant que les modifications du cristallin ont été secondaires à une inflammation sévère. Une élévation transitoire de la pression intraoculaire post-dose a été observée après les injections intravitréennes, quelle que soit la dose.
Les modifications oculaires microscopiques ont été considérées comme liées à l’inflammation et non à un processus dégénératif. Des modifications inflammatoires granulomateuses ont été observées dans la papille optique de certains yeux. Ces modifications du segment postérieur ont diminué et, dans certains cas, ont disparu, pendant la période de récupération.
Après une administration intravitréenne, aucun signe de toxicité systémique n’a été détecté. Des anticorps sériques et vitréens anti-ranibizumab ont été retrouvés chez un sous-groupe d’animaux traités.
Aucune donnée de carcinogénicité ou de mutagénicité n’est disponible.
Chez le singe, l’administration intravitréenne de ranibizumab à des femelles gestantes, ayant conduit à une exposition systémique maximale de 0,9 à 7 fois l’exposition observée en clinique, n’a pas induit de toxicité sur le développement, ni de tératogénicité, et n’a pas eu d’effet sur le poids ou la structure du placenta, bien qu’en raison de son effet pharmacologique, le ranibizumab puisse être considéré comme potentiellement tératogène et embryo/foetotoxique.
L’absence d’effets induits par le ranibizumab sur le développement embryonnaire et foetal est probablement due à l’incapacité du fragment Fab à traverser le placenta. Un cas avec des concentrations sériques élevées de ranibizumab chez la mère et la présence de ranibizumab dans le sérum foetal a toutefois été décrit, ce qui semble indiquer que les anticorps anti-ranibizumab (contenant la région Fc) ont agi comme une protéine de transport pour le ranibizumab, en diminuant ainsi son élimination du sérum maternel et en permettant son transfert placentaire. Étant donné que les études de développement embryofoetal ont été menées chez des femelles gestantes saines et que des états pathologiques (tels que le diabète) peuvent modifier la perméabilité du placenta pour le fragment Fab, les résultats de cette étude doivent être interprétés avec prudence.
INCOMPATIBILITÉS |
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver le flacon dans l’emballage extérieur, à l’abri de la lumière.
MODALITÉS MANIPULATION/ÉLIMINATION |
Flacons réservés à l’usage unique.
Pour la préparation de Lucentis pour administration intravitréenne, respecter les instructions suivantes :- Avant de prélever le contenu du flacon, la partie extérieure du bouchon en caoutchouc du flacon doit être désinfectée.
- Fixer l’aiguille-filtre de 5 µm (fournie dans la boîte) sur la seringue de 1 ml (fournie dans la boîte) en utilisant une technique aseptique. Enfoncer la pointe de l’aiguille-filtre au centre du bouchon, jusqu’à ce que l’aiguille touche le fond du flacon.
- Prélever la totalité du liquide en maintenant le flacon en position droite, légèrement inclinée pour faciliter le prélèvement complet.
- Lors du prélèvement, veiller à tirer suffisamment sur la tige du piston pour vider complètement l’aiguille-filtre.
- Laisser la pointe de l’aiguille-filtre dans le flacon et séparer la seringue de l’aiguille-filtre. L’aiguille-filtre doit être jetée après le prélèvement du contenu du flacon et ne doit pas être utilisée pour l’injection intravitréenne.
- Fixer fermement, de manière aseptique, l’aiguille pour injection (fournie dans la boîte) sur la seringue.
- Retirer avec précaution le capuchon de l’aiguille pour injection sans séparer l’aiguille de la seringue.
- Note : Tenir l’aiguille pour injection par le raccord jaune lors du retrait du capuchon.
- Expulser avec précaution l’air de la seringue et ajuster la dose au repère 0,05 ml sur la seringue. La seringue est prête pour l’injection.
- Note : Ne pas essuyer l’aiguille pour injection. Ne pas tirer à nouveau sur le piston.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Prescription réservée aux spécialistes en ophtalmologie. | |
| AMM | EU1/06/374/001 ; CIP 3400937810159 (2007, RCP rév 06.01.2011). |
| Prix : | 1093.71 euros (1 flacon). |
|
Remb Séc soc à 100 % dans le traitement de la forme néovasculaire (humide) rétrofovéolaire de la dégénérescence maculaire liée à l’âge (DMLA) selon la procédure des médicaments d’exception (prescription en conformité avec la fiche d’information thérapeutique). Le traitement par Lucentis doit être exclusivement administré par injection intravitréenne et par des ophtalmologistes expérimentés dans ce type d’injection. Non remboursable à la date du 19.01.2011 dans l’indication « Traitement de la baisse visuelle due à l’oedème maculaire diabétique (OMD) » (demande d’admission à l’étude). Collect. |
|
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne du médicament : http://www.ema.europa.eu.
Titulaire de l’AMM : Novartis Europharm Limited, Wimblehurst Road, Horsham, West Sussex, RH 125AB, Royaume-Uni.
Novartis Pharma SAS
2-4, rue Lionel-Terray. 92500 Rueil-Malmaison
Tél : 01 55 47 60 00
Information et Communication Médicales :
Tél : 01 55 47 66 00 E-mail : icm.phfr@novartis.com
Site web : http://www.novartis.fr
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


