sunitinib
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p gélule | |
| Sunitinib (DCI) malate exprimé en sunitinib | 12,5 mg |
| ou | 25 mg |
| ou | 50 mg |
INDICATIONS |
- Tumeur stromale gastro-intestinale (GIST) :
- Sutent est indiqué dans le traitement des tumeurs stromales gastro-intestinales malignes non résécables et/ou métastatiques chez l’adulte, après échec d’un traitement par le mésilate d’imatinib dû à une résistance ou à une intolérance.
- Cancer du rein métastatique (MRCC) :
- Sutent est indiqué dans le traitement des cancers du rein avancés/métastatiques (MRCC) chez l’adulte.
- Tumeur neuroendocrine du pancréas (pNET) :
- Sutent est indiqué dans le traitement des tumeurs neuroendocrines du pancréas (pNET) non résécables ou métastatiques, bien différenciées, avec progression de la maladie chez l’adulte.
POSOLOGIE ET MODE D’ADMINISTRATION |
Le traitement par le sunitinib doit être instauré par un médecin ayant l’expérience de l’administration des agents anticancéreux.
Dans les GIST et les MRCC, la dose de Sutent recommandée est de 50 mg, par voie orale, à raison d’une prise quotidienne pendant 4 semaines consécutives, suivie d’une fenêtre thérapeutique de 2 semaines (schéma posologique 4/2), correspondant à un cycle complet de 6 semaines.
Coût du traitement journalier : 135,12 euro(s).Dans les pNET, la dose de Sutent recommandée est de 37,5 mg, par voie orale, à raison d’une prise quotidienne, sans fenêtre thérapeutique préétablie.
- Ajustements de doses :
-
- Tolérance et innocuité :
Dans les GIST et les MRCC, des ajustements de doses par paliers de 12,5 mg pourront être effectués en fonction de la tolérance individuelle au traitement. La dose journalière ne devra pas excéder 75 mg ni être inférieure à 25 mg. - Dans les pNET, des ajustements de doses par paliers de 12,5 mg pourront être effectués en fonction de la tolérance individuelle au traitement. La dose maximale administrée au cours de l’étude de phase III pNET était de 50 mg par jour.
- Des interruptions de doses pourront être envisagées selon la tolérance individuelle au traitement.
- Inhibiteurs/inducteurs du CYP3A4 :
L’administration concomitante d’inducteurs puissants du CYP3A4 tels que la rifampicine devra être évitée (cf Mises en garde et Précautions d’emploi, Interactions). Si cela n’est pas possible, la dose de Sutent administrée pourra être augmentée par paliers de 12,5 mg (jusqu’à 87,5 mg par jour pour les GIST et les MRCC ou 62,5 mg par jour pour les pNET) sous étroite surveillance de la tolérance. L’administration concomitante de Sutent et d’inhibiteurs puissants du CYP3A4 tels que le kétoconazole devra être évitée (cf Mises en garde et Précautions d’emploi, Interactions). Si cela n’est pas possible, la dose de Sutent pourra être diminuée jusqu’à une dose minimale de 37,5 mg par jour pour les GIST et les MRCC ou 25 mg par jour pour les pNET, sous étroite surveillance de la tolérance. - Le choix d’un traitement médicamenteux alternatif concomitant ayant peu ou pas d’effet sur l’induction ou l’inhibition du CYP3A4 devra être envisagé.
- Tolérance et innocuité :
- Populations particulières :
-
- Population pédiatrique :
La tolérance et l’efficacité de Sutent n’ont pas été établies chez les patients de moins de 18 ans. Il n’y a pas de données disponibles. - L’utilisation de Sutent n’est pas pertinente chez l’enfant de la naissance à moins de 6 ans dans le traitement des tumeurs stromales gastro-intestinales (GIST) malignes non résécables et/ou métastatiques après échec d’un traitement par le mésilate d’imatinib dû à une résistance ou à une intolérance.
- L’utilisation de Sutent n’est pas pertinente dans la population pédiatrique dans le traitement des cancers du rein avancés/métastatiques et dans le traitement des tumeurs neuroendocrines du pancréas non résécables ou métastatiques, bien différenciées, avec progression de la maladie.
- L’utilisation de Sutent dans la population pédiatrique est déconseillée.
- Sujet âgé (>= 65 ans) :
Environ un tiers des patients ayant participé aux études cliniques et qui ont reçu le sunitinib étaient âgés de 65 ans ou plus. Aucune différence significative relative à la tolérance ou à l’efficacité n’a été observée par rapport à des patients plus jeunes. - Insuffisance hépatique :
Aucun ajustement de la dose initiale du sunitinib n’est recommandé chez les patients présentant une insuffisance hépatique légère ou modérée (classe A et B de Child-Pugh). Le sunitinib n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère de classe C de Child-Pugh (cf Pharmacocinétique). - Insuffisance rénale :
Aucun ajustement de la dose initiale de Sutent n’est exigé chez les patients présentant une altération de la fonction rénale (modérée à sévère) ou présentant une insuffisance rénale terminale (IRT) sous hémodialyse. De tels ajustements doivent être fonction de la sécurité et de la tolérance individuelle (cf Pharmacocinétique).
- Population pédiatrique :
Mode d’administration :
Sutent est destiné à une administration orale. Sutent peut être pris au cours ou en dehors d’un repas.
Si une dose est oubliée, le patient ne doit pas prendre de dose supplémentaire, mais il doit prendre la dose habituellement prescrite, le jour suivant.
CONTRE-INDICATIONS |
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- Affections de la peau et des tissus :
- Une modification de la couleur de la peau, probablement due à la couleur du principe actif (jaune), est un effet indésirable fréquent, survenant chez environ 30 % des patients. Les patients doivent également être avertis qu’une dépigmentation de la peau ou des cheveux peut également survenir pendant le traitement par le sunitinib. D’autres effets dermatologiques peuvent se produire, tels qu’une sécheresse, un épaississement ou un craquèlement de la peau, l’apparition de vésicules ou, occasionnellement, des éruptions sur la paume des mains ou la plante des pieds.
- Des douleurs ou irritations de la bouche ont été rapportées chez environ 14 % des patients.
- Les événements rapportés ci-dessus n’étaient pas cumulatifs, ils ont généralement été réversibles et en général n’ont pas nécessité l’interruption du traitement.
- Hémorragies et saignements tumoraux :
- Les événements hémorragiques, dont certains ont été d’issue fatale, rapportés après la mise sur le marché sont des hémorragies gastro-intestinales, respiratoires, tumorales, du tractus urinaire et cérébrale. Dans les essais cliniques, des hémorragies tumorales liées au traitement sont survenues chez environ 2 % des patients atteints de GIST. Elles sont susceptibles d’apparaître de façon soudaine et, dans les cas de tumeurs pulmonaires, peuvent se présenter sous forme d’hémoptysies ou d’hémorragies pulmonaires sévères mettant en jeu le pronostic vital. Des hémorragies pulmonaires mortelles sont survenues chez 2 patients (~ 1,8 %) recevant Sutent au cours d’un essai clinique de phase II portant sur des patients atteints de cancers du poumon métastatiques non à petites cellules (CPNPC). Les deux patients présentaient un carcinome épidermoïde. L’utilisation de Sutent n’est pas autorisée chez les patients atteints de CPNPC. Au cours d’une étude de phase III portant sur des patients atteints de GIST, des épisodes hémorragiques sont survenus chez 18 % des patients recevant Sutent, comparativement à 17 % des patients recevant le placebo. Parmi les patients atteints de MRCC non prétraités recevant Sutent, 39 % ont présenté des épisodes hémorragiques, comparativement à 11 % des patients recevant l’interféron-alpha (IFN-alpha). Onze patients (3,1 %) recevant du malate de sunitinib et un (0,3 %) des patients recevant l’IFN-alpha ont présenté des épisodes hémorragiques de grade 3 ou supérieur liés au traitement. Parmi les patients atteints de MRCC recevant du malate de sunitinib après l’échec d’un traitement à base de cytokine, 26 % ont présenté des épisodes hémorragiques. Au cours de l’étude de phase III portant sur des patients atteints de pNET, des épisodes hémorragiques (hors épistaxis), sont survenus chez 19 % des patients traités par le sunitinib comparativement à 4 % des patients ayant reçu un placebo. Les évaluations de routine du risque hémorragique doivent comprendre une numération formule sanguine et un examen physique.
- L’épistaxis a été l’effet indésirable hémorragique le plus fréquemment rapporté, celui-ci étant survenu chez environ la moitié des patients présentant des tumeurs solides et ayant eu des événements hémorragiques. Certains de ces épistaxis ont été sévères, mais ont très rarement évolué vers le décès.
- Affections gastro-intestinales :
- Nausée, diarrhée, stomatite, dyspepsie et vomissement ont été les effets indésirables gastro-intestinaux le plus fréquemment rapportés (cf Effets indésirables).
- La prise en charge symptomatique des effets indésirables gastro-intestinaux peut consister en un traitement par des médicaments aux propriétés antiémétiques ou antidiarrhéiques.
- Des complications gastro-intestinales graves, parfois mortelles, incluant une perforation gastro-intestinale, sont survenues dans de rares cas chez des patients présentant des tumeurs malignes intra-abdominales et traités avec le sunitinib. Une hémorragie gastro-intestinale d’issue fatale liée au traitement a été rapportée chez 0,5 % des patients recevant le placebo au cours de l’étude GIST de phase III.
- Hypertension :
- Une hypertension liée au traitement a été rapportée chez environ 16 % des patients atteints de tumeurs solides. Les doses de sunitinib ont été réduites ou son administration temporairement suspendue chez environ 2,7 % des patients qui présentèrent une hypertension. Le sunitinib n’a été définitivement arrêté chez aucun de ces patients. Une hypertension sévère (pression systolique > 200 mm Hg ou pression diastolique > 110 mm Hg) est survenue chez 4,7 % des patients présentant des tumeurs solides. Une hypertension liée au traitement a été rapportée chez environ 30 % des patients atteints de MRCC non prétraités qui recevaient le malate de sunitinib et chez 6 % de ceux qui recevaient l’IFN-alpha. Une hypertension sévère est survenue chez 12 % des patients non prétraités recevant le malate de sunitinib et chez 6 % des patients recevant l’IFN-alpha. Une hypertension liée au traitement a été rapportée chez 23 % des patients traités par le sunitinib dans l’étude de phase III pNET, comparativement à 4 % des patients ayant reçu le placebo. Une hypertension sévère est survenue chez 10 % des patients atteints de pNET traités par le sunitinib et chez 3 % des patients sous placebo. L’hypertension doit être dépistée et, si nécessaire, traitée de façon appropriée. Une interruption temporaire du traitement est recommandée chez les patients atteints d’hypertension sévère non contrôlée médicalement. Le traitement peut être repris dès que l’hypertension est correctement contrôlée.
- Affections hématologiques :
- Des diminutions du nombre absolu des neutrophiles ont été rapportées chez 10 % (grade 3) et 1,7 % (grade 4) des patients de l’étude de phase III GIST, chez 16 % (grade 3) et 1,6 % (grade 4) des patients de l’étude de phase III MRCC, et chez 13 % (grade 3) et 2,4 % (grade 4) des patients de l’étude de phase III pNET. Des diminutions du nombre des plaquettes ont été rapportées chez 3,7 % (grade 3) et 0,4 % (grade 4) des patients de l’étude de phase III GIST, chez 8,2 % (grade 3) et 1,1 % (grade 4) des patients de l’étude de phase III MRCC, et chez 3,7 % (grade 3) et 1,2 % (grade 4) des patients de l’étude de phase III pNET. Les événements rapportés ci-dessus n’étaient pas cumulatifs, et en général ont été réversibles et n’ont pas nécessité l’interruption du traitement. Aucun de ces événements pendant les études de phase III n’a été d’issue fatale, mais de rares événements hématologiques d’issue fatale, incluant des hémorragies associées à des thrombocytopénies et des infections neutropéniques, ont été rapportés après la mise sur le marché.
- Une numération formule sanguine devra être effectuée au début de chaque cycle de traitement par le sunitinib.
- Affections cardiaques :
- Les événements cardiovasculaires, incluant insuffisance cardiaque, cardiomyopathie et anomalies myocardiques, dont certains ont été d’issue fatale, ont été rapportés après la mise sur le marché. Ces données suggèrent une augmentation du risque de cardiomyopathie avec le sunitinib. A l’exception des effets spécifiques du médicament, aucun facteur de risque spécifique supplémentaire de survenue des cardiomyopathies induites par le sunitinib n’a été identifié chez les patients traités. Dans les essais cliniques, une diminution de la FEVG supérieure ou égale à 20 %, et en dessous de la limite inférieure de la normale, est survenue chez environ 2 % des patients atteints de GIST, chez 4 % des patients atteints de MRCC, traités par Sutent après échec d’un traitement à base de cytokine, et chez 2 % des patients atteints de GIST recevant un placebo. Ces diminutions de la FEVG ne semblent pas avoir été évolutives et se sont souvent améliorées avec la poursuite du traitement. Au cours de l’étude portant sur des patients ayant un MRCC non prétraité, 27 % des patients recevant Sutent et 15 % des patients recevant l’IFN-alpha ont présenté une valeur de la FEVG inférieure à la limite inférieure de la normale. Une insuffisance cardiaque congestive (ICC) a été diagnostiquée chez deux patients (moins de 1 %) ayant reçu du sunitinib.
- Chez les patients atteints de GIST, des cas d’insuffisance cardiaque, d’insuffisance cardiaque congestive ou d’insuffisance ventriculaire gauche, liés au traitement ont été rapportés chez 0,7 % des patients traités par Sutent et 1 % des patients recevant un placebo.
- Au cours de l’étude pivot de phase III chez des patients atteints de GIST (n = 312), des effets cardiaques d’issue fatale liés au traitement sont survenus chez 1 % des patients de chaque bras de l’étude (c’est-à-dire bras sunitinib et bras placebo). Au cours d’une étude de phase II chez des patients atteints de MRCC réfractaire au traitement par cytokine, un infarctus du myocarde d’issue fatale lié au traitement a été rapporté chez 0,9 % des patients et, dans l’étude de phase III, chez des patients atteints de MRCC non prétraités, des événements cardiaques d’issue fatale ont été rapportés chez 0,6 % des patients du bras IFN-alpha et aucun événement cardiaque d’issue fatale n’a été rapporté chez les patients du bras sunitinib. Dans l’étude de phase III pNET, un patient (1 %), traité par le sunitinib, a présenté une insuffisance cardiaque fatale liée au traitement. La corrélation éventuelle entre l’inhibition des récepteurs à tyrosinase kinase (RTK) et la fonction cardiaque n’est pas élucidée.
- Les patients ayant présenté des événements cardiaques dans les 12 mois précédant l’administration de sunitinib, tels qu’infarctus du myocarde (y compris angor sévère ou instable), pontage artériel coronarien ou périphérique par greffe, ICC symptomatique, accident vasculaire cérébral ou épisode ischémique transitoire, ou embolie pulmonaire, n’ont pas été inclus dans les études cliniques de Sutent. On ne sait pas si les patients atteints de ces pathologies concomitantes seraient à plus haut risque de développer une dysfonction ventriculaire gauche liée au médicament. Une surveillance stricte est recommandée chez les patients présentant des signes cliniques et symptômes d’ICC, en particulier chez les patients ayant des facteurs de risque cardiaques et/ou des antécédents de maladie coronarienne.
- Il est conseillé aux médecins de bien apprécier ce risque par rapport aux bénéfices escomptés du traitement. L’apparition de signes cliniques ou symptômes d’ICC chez ces patients devra être soigneusement surveillée au cours du traitement par le sunitinib. Des évaluations initiales et périodiques de la FEVG devront être envisagées chez les patients traités par le sunitinib. Chez les patients sans facteurs de risques cardiaques, une évaluation initiale de la fraction d’éjection devra être envisagée.
- En cas de manifestations cliniques d’ICC, il est recommandé d’interrompre l’administration de Sutent. L’administration de Sutent devra également être interrompue et/ou les doses réduites, chez les patients sans signe clinique d’ICC mais dont la fraction d’éjection a diminué de 20 % par rapport à la valeur initiale et est inférieure à 50 %.
- Allongement de l’intervalle QT :
- Les données des études non cliniques (in vitro et in vivo) menées avec des doses supérieures à la dose recommandée chez l’homme indiquent que le sunitinib peut inhiber le processus de repolarisation du potentiel d’action cardiaque (par exemple allongement de l’intervalle QT).
- Des allongements de l’intervalle QTc de plus de 500 msec se sont produits chez 0,5 % des 450 patients présentant une tumeur solide et des modifications de plus de 60 msec par rapport aux valeurs initiales chez 1,1 % de ces patients. Ces deux paramètres sont considérés comme des modifications potentiellement significatives.
- A des concentrations d’environ deux fois les concentrations thérapeutiques, le sunitinib a montré une prolongation de l’intervalle QTcF (correction de Frederica).
- L’allongement de l’intervalle QTc a été étudié dans un essai clinique, chez 24 patients âgés de 20 à 87 ans, présentant des cancers à un stade avancé. Les résultats de cette étude ont montré que le sunitinib a eu un effet sur l’intervalle QTc (défini comme une modification moyenne, ajustée pour les valeurs sous placebo, supérieure à 10 msec avec une limite supérieure de l’IC à 90 % supérieure à 15 msec) aux concentrations thérapeutiques (jour 3) en utilisant la méthode de correction des valeurs initiales en cours de journée, et aux concentrations supérieures aux concentrations thérapeutiques (jour 9) en utilisant les deux méthodes de correction des valeurs initiales. Aucun patient n’a présenté de valeur du QTc supérieure à 500 msec.
- Bien qu’un effet sur l’intervalle QTcF ait été observé 24 heures après la prise du jour 3 (concentration plasmatique thérapeutique attendue après l’administration de la dose initiale recommandée de 50 mg) avec la méthode de correction des valeurs initiales en cours de journée, la pertinence clinique de cette observation n’est pas claire.
- Les évaluations d’ECG répétées et interprétables pratiquées lors des expositions thérapeutiques ou lors d’expositions supérieures ont montré qu’aucun patient des populations évaluable ou en intention de traiter (ITT) n’avait développé un allongement de l’intervalle QTc considéré comme sévère (c’est-à-dire supérieur ou égal au grade 3 CTCAE version 3.0).
- Aux concentrations plasmatiques thérapeutiques, la modification maximale moyenne de l’intervalle QTcF (correction de Frederica) par rapport aux valeurs initiales a été de 9,6 msec (IC à 90 %, 15,1 msec). A des concentrations d’environ deux fois les concentrations thérapeutiques, la modification maximale de l’intervalle QTcF par rapport aux valeurs initiales a été de 15,4 msec (IC à 90 %, 22,4 msec). Après administration de moxifloxacine (400 mg), utilisée comme témoin positif, la modification maximale moyenne de l’intervalle QTcF par rapport aux valeurs initiales a été de 5,6 msec. Aucun sujet n’a présenté d’allongement de l’intervalle QTc supérieur au grade 2 (CTCAE version 3.0).
- L’allongement de l’intervalle QT peut entraîner une augmentation du risque d’arythmies ventriculaires y compris une torsade de pointes. Une torsade de pointes a été observée chez moins de 0,1 % des patients ayant reçu du sunitinib.
- Le sunitinib devra être utilisé avec précaution chez les patients ayant déjà présenté un allongement de l’intervalle QT, ainsi que chez ceux qui prennent des antiarythmiques ou présentant une pathologie cardiaque préexistante, une bradycardie ou des troubles électrolytiques. L’administration concomitante de sunitinib et d’inhibiteurs puissants du CYP3A4 devra être limitée en raison de la possible augmentation des concentrations plasmatiques de sunitinib (cf Posologie et Mode d’administration, Interactions).
- Effets thromboemboliques veineux :
- Des événements thromboemboliques veineux liés au traitement ont été rapportés chez environ 1,0 % des patients présentant une tumeur solide qui ont reçu Sutent dans des essais cliniques, y compris GIST et MRCC.
- Dans une étude de phase III portant sur des patients atteints de GIST, des événements thromboemboliques veineux sont survenus chez sept patients (3 %) recevant Sutent et aucun événement n’a été observé chez les patients recevant le placebo ; cinq de ces sept patients ont eu des thromboses veineuses profondes (TVP) de grade 3, et deux de ces patients des TVP de grade 1 ou 2. Quatre de ces sept patients ont vu leur traitement interrompu après la première observation de TVP.
- Des événements thromboemboliques veineux liés au traitement ont été rapportés chez treize patients (3 %) atteints de MRCC non prétraités recevant Sutent dans une étude de phase III, et chez quatre patients (2 %) inclus dans les deux études portant sur les patients atteints de MRCC après échec d’un traitement à base de cytokine. Neuf de ces patients ont présenté des embolies pulmonaires (une de grade 2 et huit de grade 4). Huit de ces patients ont présenté une TVP (une de grade 1, deux de grade 2, quatre de grade 3, et une de grade 4).
- Des événements thromboemboliques veineux sont survenus chez six patients (2 %) atteints de MRCC non prétraités recevant l’IFN-alpha : un patient (< 1 %) a présenté une TVP de grade 3 et cinq patients (1 %) des embolies pulmonaires, toutes de grade 4.
- Dans l’étude de phase III pNET, aucun événement thromboembolique veineux lié au traitement n’a été rapporté chez les patients traités par le sunitinib, et une TVP de grade 2 a été rapportée pour un patient sous placebo.
- Aucun cas d’issue fatale n’a été rapporté dans les études d’enregistrement GIST, MRCC et pNET. Des cas d’issue fatale ont été observés dans le cadre de la mise sur le marché (cf Effets respiratoires ci-dessous et Effets indésirables).
- Evénements thromboemboliques artériels :
- Des cas d’événements thromboemboliques artériels, parfois fatals, ont été rapportés chez des patients traités par le sunitinib. Les événements les plus fréquents comportaient accidents vasculaires cérébraux, accidents ischémiques transitoires et infarctus cérébraux. Les facteurs de risque associés aux événements thromboemboliques artériels, en plus de la pathologie maligne sous-jacente et de l’âge >= 65 ans, étaient l’hypertension artérielle, le diabète de type II et des antécédents de maladies thromboemboliques.
- Effets respiratoires :
- Les patients qui présentaient une embolie pulmonaire dans les 12 mois précédents les essais cliniques de Sutent ont été exclus.
- Chez les patients qui ont reçu Sutent dans les études d’enregistrement de phase III, des événements pulmonaires liés au traitement (c’est-à-dire dyspnée, épanchement pleural, embolie pulmonaire ou oedème pulmonaire) ont été rapportés chez environ 5 % des patients atteints de GIST, chez environ 14 % des patients atteints de MRCC et chez 7,2 % des patients atteints de pNET.
- Environ 8 % des patients ayant des tumeurs solides, y compris GIST et MRCC, qui ont reçu Sutent dans les essais cliniques ont présenté des événements pulmonaires liés au traitement.
- Des cas d’embolie pulmonaire ont été observés chez environ 1,3 % des patients atteints des GIST et chez environ 0,8 % des patients atteints de MRCC, qui ont reçu Sutent dans les études de phase III (cf ci-dessus Effets thromboemboliques veineux). Aucune embolie pulmonaire liée au traitement n’a été rapportée chez les patients atteints de pNET et ayant reçu le sunitinib dans l’étude de phase III. Des cas rares avec issue fatale ont été observés depuis la mise sur le marché (cf Effets indésirables).
- Dysfonction thyroïdienne :
- Une évaluation par tests biologiques de la fonction thyroïdienne préalable au traitement par le sunitinib est recommandée et les patients atteints d’hypothyroïdie ou d’hyperthyroïdie devront être traités conformément à la pratique médicale standard. Les signes et symptômes de dysfonction thyroïdienne devront être étroitement surveillés chez tous les patients traités par le sunitinib. Les patients présentant des signes et/ou des symptômes évocateurs d’une dysfonction thyroïdienne devront bénéficier d’une surveillance biologique de la fonction thyroïdienne et être traités conformément à la pratique médicale standard.
- L’hypothyroïdie a été observée aussi bien en début de traitement que tardivement au cours du traitement par le sunitinib.
- Une hypothyroïdie a été rapportée chez 7 patients (4 %) traités par Sutent inclus dans l’une ou l’autre des 2 études portant sur des patients atteints de MRCC après échec d’un traitement à base de cytokine ; chez 9 patients (2 %) recevant Sutent et chez un patient (< 1 %) recevant l’IFN-alpha dans l’étude portant sur des patients atteints de MRCC non prétraités. De plus, une élévation de la TSH a été rapportée chez 4 patients (2 %) présentant un MRCC après échec d’un traitement à base de cytokine. Globalement, 7 % des patients atteints de MRCC ont présenté des signes cliniques ou biologiques d’hypothyroïdie sous traitement. Une hypothyroïdie apparue sous traitement par Sutent a également été rapportée chez 8 patients (4 %) atteints de GIST versus 1 (1 %) sous placebo. Dans l’étude de phase III pNET, une hypothyroïdie liée au traitement a été rapportée chez 5 patients (6 %) traités par le sunitinib, et chez un patient (1 %) sous placebo.
- De rares cas d’hyperthyroïdie, parfois suivie d’une hypothyroïdie, ont été rapportés dans les essais cliniques et après la mise sur le marché.
- Pancréatite :
- Des augmentations sériques des lipases et des amylases ont été observées chez des patients présentant diverses formes de tumeurs solides et ayant reçu du sunitinib. Les augmentations sériques des lipases ont été transitoires et généralement non associées à des signes ou symptômes de pancréatite chez des patients présentant diverses formes de tumeurs solides. Un cas de pancréatite a été peu fréquemment (< 1 %) observé chez les patients atteints de MRCC ou de GIST et traités par Sutent. Des cas graves de pancréatite, dont certains d’issue fatale, ont été rapportés.
- En présence de symptômes de pancréatite, le traitement par le sunitinib devrait être arrêté et les patients devront bénéficier d’un suivi médical approprié.
- Dans l’étude de phase III pNET, aucun cas de pancréatite liée au traitement n’a été rapporté.
- Hépatotoxicité :
- Une hépatotoxicité a été observée chez des patients traités par le sunitinib. Des cas d’insuffisance hépatique, dont certains d’issue fatale, ont été observés chez < 1 % des patients présentant une tumeur solide et traités par le sunitinib. Surveiller les tests de la fonction hépatique (taux d’alanine aminotransférase [ALAT], d’aspartate aminotransférase [ASAT], de bilirubine) avant l’initiation du traitement, au cours de chaque cycle de traitement, et en cas de symptômes cliniques.
- En présence de signes ou symptômes d’insuffisance hépatique, le traitement par Sutent doit être arrêté et un traitement approprié doit être mis en place.
- Fonction rénale :
- Des cas d’altération de la fonction rénale, d’insuffisance rénale et/ou d’insuffisance rénale aiguë, dont certains d’issue fatale, ont été rapportés.
- En plus du carcinome rénal sous-jacent, les facteurs de risque associés à l’altération de la fonction rénale/l’insuffisance rénale chez les patients traités par le sunitinib étaient les suivants : patients plus âgés, diabète de type II, insuffisance rénale sous-jacente, insuffisance cardiaque, hypertension, sepsis, déshydratation/hypovolémie et rhabdomyolyse.
- La sécurité des patients présentant une protéinurie modérée à sévère, poursuivant le traitement par Sutent, n’a pas été systématiquement évaluée.
- Des cas de protéinuries et de rares cas de syndrome néphrotique ont été rapportés.
- Il est recommandé de pratiquer une analyse urinaire avant l’initiation du traitement et de surveiller l’apparition ou l’aggravation d’une protéinurie. Le traitement par Sutent doit être arrêté chez les patients présentant un syndrome néphrotique.
- Fistule :
- Si la formation d’une fistule se produit, le traitement par le sunitinib doit être interrompu. Peu d’informations sont disponibles sur la poursuite de l’utilisation de sunitinib chez des patients présentant des fistules.
- Troubles de la cicatrisation des plaies :
- Des cas de troubles de la cicatrisation des plaies ont été rapportés au cours du traitement par le sunitinib.
- Aucune étude clinique formelle de l’effet du sunitinib sur la cicatrisation des plaies n’a été menée. Par précaution, une interruption temporaire du traitement est recommandée chez les patients devant subir une intervention chirurgicale majeure. L’expérience clinique concernant le délai de réintroduction du traitement après une intervention chirurgicale majeure est limitée. Aussi, la décision de reprendre le traitement par le sunitinib après une intervention chirurgicale majeure doit se baser sur l’appréciation clinique du rétablissement après la chirurgie.
- Ostéonécrose de la mâchoire :
- Des cas d’ostéonécrose de la mâchoire ont été rapportés chez des patients traités par Sutent. La majorité de ces cas est survenue chez des patients ayant reçu antérieurement ou de façon concomitante un traitement par des biphosphonates par voie IV, pour lequel l’ostéonécrose de la mâchoire est un risque identifié. La prudence est donc de rigueur chez les patients traités par Sutent en cas d’administration antérieure ou concomitante de biphosphonates par voie IV.
- Les interventions dentaires invasives sont également un facteur de risque identifié. Avant d’instaurer un traitement par Sutent, un examen dentaire et des soins dentaires préventifs appropriés doivent être envisagés. Les interventions dentaires invasives doivent être évitées autant que possible chez les patients qui ont reçu précédemment ou qui reçoivent des biphosphonates par voie IV (cf Effets indésirables).
- Hypersensibilité/angio-oedème :
- Si un angio-oedème dû à de l’hypersensibilité se produit, le traitement par le sunitinib doit être interrompu et le traitement médical standard doit être appliqué.
- Affections du système nerveux :
- Troubles du goût. Une dysgueusie a été rapportée chez environ 28 % des patients traités par Sutent dans les études cliniques.
- Crises convulsives :
- Dans les études cliniques et après la mise sur le marché de Sutent, certains patients avec ou sans signes radiologiques de métastases cérébrales ont présenté des crises convulsives. En outre, peu de cas de convulsions (< 1 %) associées à des signes radiologiques d’un syndrome de leuco-encéphalopathie postérieure réversible (RPLS) ont été rapportés. Les patients atteints de crises convulsives et présentant des signes/symptômes suggérant un RPLS, tels qu’hypertension, céphalées, baisse de vigilance ou des facultés mentales, perte de la vision, et notamment cécité corticale, devront être surveillés et traités pour leur hypertension. Il est recommandé d’interrompre temporairement la prise de Sutent ; après normalisation de l’état du patient, le traitement pourra être repris selon l’avis du médecin.
INTERACTIONS |
Les études d’interaction ont été menées uniquement chez l’adulte.
- Médicaments pouvant augmenter les concentrations plasmatiques de sunitinib :
Chez les volontaires sains, l’administration concomitante d’une dose unique de sunitinib et d’un puissant inhibiteur du CYP3A4, le kétoconazole, a provoqué une élévation de la valeur de la Cmax et de la valeur de l’AUC0-infini de la combinaison (sunitinib + métabolite principal) de 49 % et 51 % respectivement. - L’administration concomitante de sunitinib et d’inhibiteurs puissants du CYP3A4 (tels que le ritonavir, l’itraconazole, l’érythromycine, la clarithromycine, le jus de pamplemousse) peut augmenter les concentrations de sunitinib.
- L’association de Sutent avec des inhibiteurs du CYP3A4 devra donc être évitée, ou l’utilisation d’un autre traitement pris de façon concomitante et présentant un potentiel inhibiteur du CYP3A4 minimal ou nul devra être envisagée.
- Si cela n’est pas possible, la dose de Sutent pourra être réduite jusqu’à une dose minimale journalière de 37,5 mg pour les GIST et MRCC ou de 25 mg pour les pNET, sous surveillance étroite de la tolérance (cf Posologie et Mode d’administration).
- Médicaments pouvant diminuer les concentrations plasmatiques de sunitinib :
Chez les volontaires sains, l’administration concomitante d’une dose unique de sunitinib et d’un inducteur du CYP3A4, la rifampicine, a provoqué une diminution de la valeur de la Cmax et de la valeur de l’AUC0-infini de la combinaison (sunitinib + métabolite principal) de 23 % et 46 % respectivement. - L’administration concomitante de sunitinib et d’inducteurs puissants du CYP3A4 (tels que la dexaméthasone, la phénytoïne, la carbamazépine, la rifampicine, le phénobarbital ou des préparations à base de plante contenant de l’Hypericum perforatum [millepertuis]) peut diminuer les concentrations de sunitinib. L’association de sunitinib avec des inducteurs du CYP3A4 devra donc être évitée, ou le choix d’un autre traitement concomitant ayant un potentiel inducteur sur le CYP3A4 nul ou réduit devra être envisagé.
- Si cela n’est pas possible, la dose de Sutent pourra être augmentée par paliers de 12,5 mg (jusqu’à une dose maximale journalière de 87,5 mg pour les GIST et les MRCC ou de 62,5 mg par jour pour les pNET), sous surveillance étroite de la tolérance (cf Posologie et Mode d’administration).
- Anticoagulants : de rares cas d’hémorragies ont été observés chez des patients traités par le sunitinib (cf Mises en garde et Précautions d’emploi, Effets indésirables). Les patients recevant un traitement anticoagulant concomitant (par exemple : warfarine ou acénocoumarol) pourront être surveillés de façon périodique en procédant à des numérations formule sanguine (plaquettes), des tests de facteurs de coagulation (TP/INR) et des examens physiques.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Pour Sutent on ne dispose d’aucune étude chez la femme enceinte. Les études animales ont mis en évidence une toxicité sur la reproduction, comprenant des malformations foetales (cf Sécurité préclinique). Sutent ne doit pas être utilisé pendant la grossesse ou chez des femmes n’utilisant pas de méthode de contraception efficace, à moins que les bénéfices escomptés ne justifient le risque potentiel pour le foetus. Si Sutent est utilisé pendant la grossesse ou si la patiente devient enceinte en cours de traitement par Sutent, elle devra être avertie des risques potentiels pour le foetus.
Il est conseillé aux femmes en âge de procréer d’utiliser une méthode de contraception efficace et d’éviter d’être enceintes au cours du traitement par Sutent.
Allaitement :
Le sunitinib et/ou ses métabolites sont excrétés dans le lait du rat femelle. On ne sait pas si le sunitinib ou son métabolite actif principal est excrété dans le lait maternel de la femme. Dans la mesure où les substances actives sont généralement excrétées dans le lait maternel et où il existe un risque potentiel d’événements indésirables graves chez le nouveau-né, les femmes ne doivent pas allaiter pendant un traitement par Sutent.
Fécondité :Selon des données non cliniques, la fertilité des hommes et des femmes pourrait être affectée par le traitement par Sutent (cf Sécurité préclinique).
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
| Système organe/classe | |||
| Fréquence : – Effets indésirables | Tout grade n (%) | Grade 3 n (%) | Grade 4 n (%) |
| Affections hématologiques et du système lymphatique : | |||
| Très fréquent : | |||
| – Anémie | 86 (19,5) | 24 (5,5) | 3 (0,7) |
| – Neutropénie | 81 (18,4) | 39 (8,9) | 5 (1,1) |
| – Thrombocytopénie | 67 (15,2) | 19 (4,3) | 6 (1,4) |
| Fréquent : | |||
| – Leucopénie | 26 (5,9) | 9 (2,0) | 1 (0,2) |
| – Lymphopénie | 10 (2,3) | 3 (0,7) | 1 (0,2) |
| Affections endocriniennes : | |||
| Très fréquent : | |||
| – Hypothyroïdie | 59 (13,4) | 5 (1,1) | 1 (0,2) |
| Troubles du métabolisme et de la nutrition : | |||
| Très fréquent : | |||
| – Diminution de l’appétit(a)
| 117 (26,6) | 8 (1,8) | 0 (0,0) |
| Affections psychiatriques : | |||
| Fréquent : | |||
| – Insomnie | 14 (3,2) | 0 (0,0) | 0 (0,0) |
| Affections du système nerveux : | |||
| Très fréquent : | |||
| – Troubles du goût(b)
| 105 (23,9) | 1 (0,2) | 0 (0,0) |
| – Céphalées | 76 (17,3) | 5 (1,1) | 0 (0,0) |
| Fréquent : | |||
| – Paresthésies | 27 (6,1) | 1 (0,2) | 0 (0,0) |
| – Sensation vertigineuse | 18 (4,1) | 1 (0,2) | 0 (0,0) |
| – Neuropathie périphérique | 11 (2,5) | 0 (0,0) | 0 (0,0) |
| – Hypoesthésie | 10 (2,3) | 0 (0,0) | 0 (0,0) |
| Affections vasculaires : | |||
| Très fréquent : | |||
| – Hypertension | 101 (23,0) | 43 (9,8) | 0 (0,0) |
| Affections respiratoires, thoraciques et médiastinales : | |||
| Fréquent : | |||
| – Épistaxis | 28 (6,4) | 1 (0,2) | 0 (0,0) |
| – Dyspnée | 16 (3,6) | 2 (0,5) | 0 (0,0) |
| Affections du rein et des voies urinaires : | |||
| Fréquent : | |||
| – Chromaturie | 18 (4,1) | 0 (0,0) | 0 (0,0) |
| Affections gastro-intestinales : | |||
| Très fréquent : | |||
| – Diarrhée | 187 (42,5) | 24 (5,5) | 0 (0,0) |
| – Nausée | 161 (36,6) | 15 (3,4) | 0 (0,0) |
| – Stomatite | 90 (20,5) | 7 (1,6) | 0 (0,0) |
| – Vomissements | 98 (22,2) | 7 (1,6) | 0 (0,0) |
| – Dyspepsie | 80 (18,2) | 4 (0,9) | 0 (0,0) |
| – Douleur abdominale(c)/Distension | 77 (17,5) | 15 (3,4) | 2 (0,5) |
| – Flatulence | 46 (10,5) | 0 (0,0) | 0 (0,0) |
| – Douleur buccale | 44 (10,0) | 2 (0,5) | 0 (0,0) |
| Fréquent : | |||
| – Glossodynie | 37 (8,4) | 0 (0,0) | 0 (0,0) |
| – Constipation | 37 (8,4) | 2 (0,5) | 0 (0,0) |
| – Sécheresse de la bouche | 31 (7,0) | 0 (0,0) | 0 (0,0) |
| – Reflux gastro-oesophagien | 12 (2,7) | 1 (0,2) | 0 (0,0) |
| – Aphte buccal | 11 (2,5) | 0 (0,0) | 0 (0,0) |
| – Gêne buccale | 11 (2,5) | 0 (0,0) | 0 (0,0) |
| Affections de la peau et du tissu sous-cutané : | |||
| Très fréquent : | |||
| – Coloration jaune de la peau/Modification de la couleur de la peau | 146 (33,2) | 0 (0,0) | 0 (0,0) |
| – Érythrodysesthésie palmoplantaire | 106 (24,1) | 27 (6,1) | 0 (0,0) |
| – Rash | 64 (14,5) | 3 (0,7) | 0 (0,0) |
| – Modification de la couleur des cheveux | 67 (15,2) | 0 (0,0) | 0 (0,0) |
| Fréquent : | |||
| – Sécheresse de la peau | 41 (9,3) | 0 (0,0) | 0 (0,0) |
| – Alopécie | 33 (7,5) | 0 (0,0) | 0 (0,0) |
| – Dermatite | 29 (6,6) | 1 (0,2) | 0 (0,0) |
| – OEdème périorbital | 20 (4,5) | 0 (0,0) | 0 (0,0) |
| – Réaction cutanée | 20 (4,5) | 3 (0,7) | 0 (0,0) |
| – Érythème | 18 (4,1) | 0 (0,0) | 0 (0,0) |
| – Eczéma | 16 (3,6) | 1 (0,2) | 0 (0,0) |
| – Prurit | 16 (3,6) | 0 (0,0) | 0 (0,0) |
| – Hyperpigmentation de la peau | 15 (3,4) | 0 (0,0) | 0 (0,0) |
| – Exfoliation cutanée | 12 (2,7) | 0 (0,0) | 0 (0,0) |
| – Ampoule | 10 (2,3) | 1 (0,2) | 0 (0,0) |
| – Lésion cutanée | 10 (2,3) | 1 (0,2) | 0 (0,0) |
| Affections musculosquelettiques et systémiques : | |||
| Très fréquent : | |||
| – Douleurs des extrémités/des membres | 54 (12,3) | 5 (1,1) | 0 (0,0) |
| Fréquent : | |||
| – Arthralgie | 39 (8,9) | 3 (0,7) | 0 (0,0) |
| – Myalgie | 29 (6,6) | 0 (0,0) | 0 (0,0) |
| – Spasme musculaire | 21 (4,8) | 1 (0,2) | 0 (0,0) |
| – Dorsalgie | 11 (2,5) | 2 (0,5) | 0 (0,0) |
| – Faiblesse musculaire | 10 (2,3) | 1 (0,2) | 0 (0,0) |
| Troubles généraux et anomalies au site d’administration : | |||
| Très fréquent : | |||
| – Fatigue/Asthénie | 287 (65,2) | 64 (14,5) | 5 (1,1) |
| – Inflammation des muqueuses | 70 (15,9) | 6 (1,4) | 1 (0,2) |
| – OEdème(d)
| 59 (13,4) | 1 (0,2) | 0 (0,0) |
| Fréquent : | |||
| – Pyrexie | 26 (5,9) | 2 (0,5) | 0 (0,0) |
| Investigations : | |||
| Fréquent : | |||
| – Baisse du taux d’hémoglobine | 27 (6,1) | 6 (1,4) | 0 (0,0) |
| – Diminution des globules blancs(e)
| 33 (7,5) | 15 (3,4) | 0 (0,0) |
| – Élévation de la créatinine-phosphokinase sérique | 22 (5,0) | 1 (0,2) | 1 (0,2) |
| – Diminution de la fraction d’éjection | 27 (6,1) | 5 (1,2) | 0 (0,0) |
| – Élévation de la lipase | 35 (8,0) | 12 (2,7) | 7 (1,6) |
| – Baisse du nombre des plaquettes | 25 (5,7) | 4 (0,9) | 1 (0,2) |
| – Perte de poids | 23 (5,2) | 1 (0,2) | 0 (0,0) |
| – Élévation de l’amylase | 21 (4,8) | 8 (1,8) | 0 (0,0) |
| – Élévation de l’aspartate aminotransférase | 18 (4,1) | 2 (0,5) | 1 (0,2) |
| – Élévation de l’alanine aminotransférase | 12 (2,7) | 1 (0,2) | 0 (0,0) |
| Tout effet indésirable | 414 (94,1) | 204 (46,4) | 53 (12,0) |
(b) dysgueusie, agueusie et troubles du goût
(c) douleurs abdominales et douleurs abdominales hautes
(d) oedème, oedème périphérique et oedème du visage
(e) diminution des globules blancs, diminution des neutrophiles et diminution des leucocytes
| Système organe/classe | |||
| Fréquence : – Effets indésirables | Tout grade n (%) | Grade 3 n (%) | Grade 4 n (%) |
| Affections hématologiques et du système lymphatique : | |||
| Très fréquent : | |||
| – Neutropénie | 89 (16,4) | 46 (8,5) | 5 (0,9) |
| – Thrombocytopénie | 86 (15,8) | 37 (6,8) | 5 (0,9) |
| – Anémie | 68 (12,5) | 21 (3,9) | 4 (0,7) |
| – Leucopénie | 55 (10,1) | 20 (3,7) | 0 (0,0) |
| Fréquent : | |||
| – Lymphopénie | 21 (3,9) | 12 (2,2) | 1 (0,2) |
| Affections endocriniennes : | |||
| Très fréquent : | |||
| – Hypothyroïdie | 69 (12,7) | 7 (1,3) | 0 (0,0) |
| Troubles du métabolisme et de la nutrition : | |||
| Très fréquent : | |||
| – Diminution de l’appétit(a)
| 205 (37,7) | 9 (1,7) | 0 (0,0) |
| Fréquent : | |||
| – Déshydratation | 33 (6,1) | 7 (1,3) | 1 (0,2) |
| Affections psychiatriques : | |||
| Fréquent : | |||
| – Insomnie | 22 (4,0) | 0 (0,0) | 0 (0,0) |
| – Dépression | 15 (2,8) | 1 (0,2) | 0 (0,0) |
| Affections du système nerveux : | |||
| Très fréquent : | |||
| – Troubles du goût(b)
| 251 (46,1) | 1 (0,2) | 0 (0,0) |
| – Céphalées | 82 (15,1) | 3 (0,6) | 0 (0,0) |
| Fréquent : | |||
| – Sensations vertigineuses | 38 (7,0) | 2 (0,4) | 0 (0,0) |
| – Neuropathie périphérique | 35 (6,4) | 2 (0,4) | 0 (0,0) |
| – Paresthésie | 35 (6,4) | 0 (0,0) | 0 (0,0) |
| – Hypoesthésie | 20 (3,7) | 0 (0,0) | 0 (0,0) |
| – Hyperesthésie | 18 (3,3) | 0 (0,0) | 0 (0,0) |
| Affections oculaires : | |||
| Fréquent : | |||
| – Augmentation des sécrétions lacrymales | 39 (7,2) | 0 (0,0) | 0 (0,0) |
| – OEdème des paupières | 12 (2,2) | 0 (0,0) | 0 (0,0) |
| Affections vasculaires : | |||
| Très fréquent : | |||
| – Hypertension | 149 (27,4) | 56 (10,3) | 0 (0,0) |
| Fréquent : | |||
| – Bouffée vasomotrice | 17 (3,1) | 0 (0,0) | 0 (0,0) |
| – Bouffée de chaleur | 12 (2,2) | 0 (0,0) | 0 (0,0) |
| Affections respiratoires, thoraciques et médiastinales : | |||
| Très fréquent : | |||
| – Épistaxis | 87 (16,0) | 3 (0,6) | 0 (0,0) |
| Fréquent : | |||
| – Dyspnée | 45 (8,3) | 6 (1,1) | 0 (0,0) |
| – Douleur laryngopharyngée | 26 (4,8) | 2 (0,4) | 0 (0,0) |
| – Toux | 23 (4,2) | 0 (0,0) | 0 (0,0) |
| – Dysphonie | 16 (2,9) | 0 (0,0) | 0 (0,0) |
| – Sécheresse nasale | 14 (2,6) | 0 (0,0) | 0 (0,0) |
| – Épanchement pleural | 12 (2,2) | 3 (0,6) | 0 (0,0) |
| – Congestion nasale | 12 (2,2) | 0 (0,0) | 0 (0,0) |
| – Dyspnée d’effort | 11 (2,0) | 0 (0,0) | 0 (0,0) |
| Affections gastro-intestinales : | |||
| Très fréquent : | |||
| – Diarrhée | 326 (59,9) | 39 (7,2) | 0 (0,0) |
| – Nausée | 290 (53,3) | 19 (3,5) | 0 (0,0) |
| – Stomatite/Stomatite aphteuse | 192 (35,3) | 14 (2,6) | 0 (0,0) |
| – Dyspepsie | 189 (34,7) | 8 (1,5) | 0 (0,0) |
| – Vomissements | 180 (33,1) | 17 (3,1) | 0 (0,0) |
| – Douleur abdominale(c)/Distension | 99 (18,2) | 9 (1,7) | 0 (0,0) |
| – Constipation | 83 (15,3) | 1 (0,2) | 0 (0,0) |
| – Glossodynie | 63 (11,6) | 0 (0,0) | 0 (0,0) |
| – Douleur buccale | 62 (11,4) | 2 (0,4) | 0 (0,0) |
| – Flatulence | 60 (11,0) | 0 (0,0) | 0 (0,0) |
| – Sécheresse de la bouche | 56 (10,3) | 0 (0,0) | 0 (0,0) |
| Fréquent : | |||
| – Reflux gastro-oesophagien | 50 (9,2) | 2 (0,4) | 0 (0,0) |
| – Dysphagie | 20 (3,7) | 2 (0,4) | 1 (0,2) |
| – Chéilite | 19 (3,5) | 1 (0,2) | 0 (0,0) |
| – Saignement gingival | 18 (3,3) | 0 (0,0) | 0 (0,0) |
| – Hémorroïdes | 18 (3,3) | 0 (0,0) | 0 (0,0) |
| – Proctalgie | 17 (3,1) | 1 (0,2) | 0 (0,0) |
| – Aphte buccal | 16 (2,9) | 0 (0,0) | 1 (0,2) |
| – Hémorragie rectale | 13 (2,4) | 0 (0,0) | 0 (0,0) |
| – Gêne gastrique | 12 (2,2) | 0 (0,0) | 0 (0,0) |
| – Éructation | 11 (2,0) | 0 (0,0) | 0 (0,0) |
| Affections de la peau et du tissu sous-cutané : | |||
| Très fréquent : | |||
| – Coloration jaune de la peau/Modification de la couleur de la peau/Troubles de la pigmentation | 153 (28,1) | 1 (0,2) | 0 (0,0) |
| – Érythrodysesthésie palmoplantaire | 139 (25,6) | 44 (8,1) | 0 (0,0) |
| – Rash | 122 (22,4) | 3 (0,6) | 1 (0,2) |
| – Sécheresse de la peau | 108 (19,9) | 1 (0,2) | 0 (0,0) |
| – Modification de la couleur des cheveux | 103 (18,9) | 0 (0,0) | 0 (0,0) |
| – Alopécie | 64 (11,8) | 0 (0,0) | 0 (0,0) |
| – Érythème | 58 (10,7) | 2 (0,4) | 0 (0,0) |
| Fréquent : | |||
| – Dermatite exfoliative | 47 (8,6) | 4 (0,7) | 0 (0,0) |
| – Réaction cutanée/Troubles cutanés | 42 (7,7) | 6 (1,1) | 0 (0,0) |
| – Prurit | 40 (7,4) | 1 (0,2) | 0 (0,0) |
| – OEdème périorbital | 31 (5,7) | 1 (0,2) | 0 (0,0) |
| – Lésion cutanée | 27 (5,0) | 1 (0,2) | 0 (0,0) |
| – Dermatite | 26 (4,8) | 4 (0,7) | 0 (0,0) |
| – Altération des ongles/Modification de la couleur | 25 (4,6) | 0 (0,0) | 0 (0,0) |
| – Ampoule | 23 (4,2) | 1 (0,2) | 0 (0,0) |
| – Hyperkératose | 22 (4,0) | 4 (0,7) | 0 (0,0) |
| – Acné | 19 (3,5) | 0 (0,0) | 0 (0,0) |
| Affections musculosquelettiques et systémiques : | |||
| Très fréquent : | |||
| – Douleurs des extrémités | 96 (17,6) | 6 (1,1) | 0 (0,0) |
| Fréquent : | |||
| – Arthralgie | 51 (9,4) | 1 (0,2) | 0 (0,0) |
| – Myalgie | 49 (9,0) | 2 (0,4) | 0 (0,0) |
| – Spasme musculaire | 26 (4,8) | 0 (0,0) | 0 (0,0) |
| – Dorsalgie | 17 (3,1) | 2 (0,4) | 0 (0,0) |
| – Douleur musculosquelettique | 11 (2,0) | 2 (0,4) | 0 (0,0) |
| Affections du rein et des voies urinaires : | |||
| Fréquent : | |||
| – Chromaturie | 17 (3,1) | 0 (0,0) | 0 (0,0) |
| Troubles généraux et anomalies au site d’administration : | |||
| Très fréquent : | |||
| – Fatigue/Asthénie | 373 (68,6) | 93 (17,1) | 1 (0,2) |
| – Inflammation des muqueuses | 134 (24,6) | 8 (1,5) | 0 (0,0) |
| – OEdème(d)
| 83 (15,3) | 4 (0,7) | 0 (0,0) |
| Fréquent : | |||
| – Fièvre | 37 (6,8) | 3 (0,6) | 0 (0,0) |
| – Frissons | 34 (6,3) | 2 (0,4) | 0 (0,0) |
| – Douleur | 21 (3,9) | 0 (0,0) | 0 (0,0) |
| – Douleur thoracique | 13 (2,4) | 2 (0,4) | 0 (0,0) |
| – Syndrome pseudogrippal | 11 (2,0) | 0 (0,0) | 0 (0,0) |
| Investigations : | |||
| Très fréquent : | |||
| – Fraction d’éjection diminuée/anormale | 86 (15,8) | 16 (2,9) | 0 (0,0) |
| – Perte de poids | 58 (10,7) | 1 (0,2) | 0 (0,0) |
| Fréquent : | |||
| – Diminution du nombre des plaquettes | 41 (7,5) | 15 (2,8) | 2 (0,4) |
| – Diminution des globules blancs(e)
| 37 (6,8) | 16 (2,9) | 0 (0,0) |
| – Élévation de la lipase | 36 (6,6) | 19 (3,5) | 11 (2,0) |
| – Diminution de l’hémoglobine | 25 (4,6) | 8 (1,5) | 0 (0,0) |
| – Élévation de l’amylasémie | 19 (3,5) | 11 (2,0) | 2 (0,4) |
| – Élévation sanguine des créatines phosphokinases | 19 (3,5) | 7 (1,3) | 2 (0,4) |
| – Élévation de l’aspartate aminotransférase | 18 (3,3) | 7 (1,3) | 0 (0,0) |
| – Élévation de la créatininémie | 17 (3,1) | 3 (0,6) | 0 (0,0) |
| – Augmentation de la tension artérielle | 15 (2,8) | 2 (0,4) | 0 (0,0) |
| – Élévation de l’alanine aminotransférase | 14 (2,6) | 7 (1,3) | 2 (0,4) |
| Tout effet indésirable | 524 (96,3) | 297 (54,6) | 59 (10,8) |
(b) dysgueusie, agueusie et troubles du goût
(c) douleurs abdominales et douleurs abdominales hautes
(d) oedème, oedème périphérique et oedème du visage
(e) diminution des globules blancs, diminution des neutrophiles et diminution des leucocytes
| Système organe/classe | |||
| Fréquence : – Effets indésirables | Tout grade n (%) | Grade 3 n (%) | Grade 4 n (%) |
| Affections hématologiques et du système lymphatique : | |||
| Très fréquent : | |||
| – Neutropénie | 24 (28,9) | 6 (7,2) | 4 (4,8) |
| – Thrombocytopénie | 14 (16,9) | 2 (2,4) | 1 (1,2) |
| Fréquent : | |||
| – Leucopénie | 8 (9,6) | 4 (4,8) | 1 (1,2) |
| Affections endocriniennes : | |||
| Fréquent : | |||
| – Hypothyroïdie | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| Troubles métaboliques et de la nutrition : | |||
| Très fréquent : | |||
| – Anorexie | 17 (20,5) | 2 (2,4) | 0 (0,0) |
| Fréquent : | |||
| – Diminution de l’appétit | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| Affections psychiatriques : | |||
| Fréquent : | |||
| – Insomnie | 7 (8,4) | 0 (0,0) | 0 (0,0) |
| Affections du système nerveux : | |||
| Très fréquent : | |||
| – Dysgueusie | 16 (19,3) | 0 (0,0) | 0 (0,0) |
| – Céphalées | 10 (12,0) | 0 (0,0) | 0 (0,0) |
| Fréquent : | |||
| – Sensations vertigineuses | 5 (6,0) | 1 (1,2) | 0 (0,0) |
| Affections oculaires : | |||
| Fréquent : | |||
| – OEdème des paupières | 5 (6,0) | 1 (1,2) | 0 (0,0) |
| Affections vasculaires : | |||
| Très fréquent : | |||
| – Hypertension | 19 (22,9) | 8 (9,6) | 0 (0,0) |
| Affections respiratoires, thoraciques et médiastinales : | |||
| Très fréquent : | |||
| – Épistaxis | 16 (19,3) | 1 (1,2) | 0 (0,0) |
| Fréquent : | |||
| – Dyspnée | 6 (7,2) | 1 (1,2) | 0 (0,0) |
| Affections gastro-intestinales : | |||
| Très fréquent : | |||
| – Diarrhée | 44 (53,0) | 4 (4,8) | 0 (0,0) |
| – Nausée | 32 (38,6) | 1 (1,2) | 0 (0,0) |
| – Vomissements | 21 (25,3) | 0 (0,0) | 0 (0,0) |
| – Stomatite | 18 (21,7) | 3 (3,6) | 0 (0,0) |
| – Douleur abdominale | 12 (14,5) | 1 (1,2) | 0 (0,0) |
| – Dyspepsie | 12 (14,5) | 0 (0,0) | 0 (0,0) |
| Fréquent : | |||
| – Constipation | 8 (9,6) | 0 (0,0) | 0 (0,0) |
| – Sécheresse de la bouche | 7 (8,4) | 0 (0,0) | 0 (0,0) |
| – Douleur abdominale haute | 6 (7,2) | 1 (1,2) | 0 (0,0) |
| – Stomatite aphteuse | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| – Flatulence | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| – Saignement gingival | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| Affections de la peau et du tissu sous-cutané : | |||
| Très fréquent : | |||
| – Modification de la couleur des cheveux | 24 (28,9) | 1 (1,2) | 0 (0,0) |
| – Érythrodysesthésie palmoplantaire | 19 (22,9) | 5 (6,0) | 0 (0,0) |
| – Rash | 13 (15,7) | 0 (0,0) | 0 (0,0) |
| – Sécheresse de la peau | 11 (13,3) | 0 (0,0) | 0 (0,0) |
| Fréquent : | |||
| – Altération des ongles | 8 (9,6) | 0 (0,0) | 0 (0,0) |
| – Érythème | 7 (8,4) | 0 (0,0) | 0 (0,0) |
| – Coloration jaune de la peau | 6 (7,2) | 0 (0,0) | 0 (0,0) |
| – Alopécie | 5 (6,0) | 0 (0,0) | 0 (0,0) |
| Affections musculosquelettiques et systémiques : | |||
| Fréquent : | |||
| – Douleur aux extrémités | 7 (8,4) | 0 (0,0) | 0 (0,0) |
| – Arthralgie | 6 (7,2) | 0 (0,0) | 0 (0,0) |
| Troubles généraux et anomalies au site d’administration : | |||
| Très fréquent : | |||
| – Fatigue/Asthénie | 46 (55,4) | 5 (6,0) | 1 (1,2) |
| – Inflammation des muqueuses | 13 (15,7) | 1 (1,2) | 0 (0,0) |
| – Perte de poids | 11 (13,3) | 1 (1,2) | 0 (0,0) |
| Tout effet indésirable : | 81 (97,6) | 29 (34,9) | 7 (8,4) |
- Effets indésirables rapportés depuis la mise sur le marché :
- Les types d’effets suivants ont été rapportés après la mise sur le marché de Sutent. Ce sont des cas notifiés de façon spontanée ainsi que des événements indésirables graves survenus dans les études en cours, les programmes d’accès étendus, les études de pharmacologie clinique et les études exploratoires dans des indications non approuvées.
- Infections et infestations :
- Fréquence indéterminée : infections (avec ou sans neutropénie associée).
- Des cas graves d’infection (avec ou sans neutropénie associée), y compris pneumonie, ont été rapportés. Quelques cas ont été d’issue fatale.
- Fréquence indéterminée : infections (avec ou sans neutropénie associée).
- Affections hématologiques et du système lymphatique :
- Fréquence indéterminée : microangiopathie thrombotique.
- De rares cas de microangiopathie thrombotique ont été rapportés. L’arrêt temporaire de Sutent est recommandé ; après résolution et à l’appréciation du médecin, le traitement pourra être repris.
- Fréquence indéterminée : microangiopathie thrombotique.
- Affections du système immunitaire :
- Fréquence indéterminée : angio-oedème, réaction d’hypersensibilité
Des réactions d’hypersensibilité ont été rapportées, incluant des angio-oedèmes.
- Fréquence indéterminée : angio-oedème, réaction d’hypersensibilité
- Affections endocriniennes :
- Fréquence indéterminée : hyperthyroïdie.
- De rares cas d’hyperthyroïdies, dont certaines suivies d’une hypothyroïdie, ont été rapportés dans les essais cliniques et après la mise sur le marché (cf Mises en garde et Précautions d’emploi également).
- Fréquence indéterminée : hyperthyroïdie.
- Affections cardiaques :
- Peu fréquent : insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance ventriculaire gauche.
- Rare : allongement de l’intervalle QT, torsades de pointes.
- Fréquence indéterminée : cardiomyopathie, épanchement péricardique.
- Peu fréquent : insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance ventriculaire gauche.
- Affections gastro-intestinales :
- Peu fréquent : pancréatite.
- Rare : perforation gastro-intestinale.
- Peu fréquent : pancréatite.
- Affections hépatobiliaires :
- Peu fréquent : insuffisance hépatique.
- Fréquence indéterminée : hépatite.
- Des troubles de la fonction hépatique ont été rapportés et peuvent inclure des anomalies des tests de la fonction hépatique, des hépatites ou des insuffisances hépatiques (cf également Mises en garde et Précautions d’emploi).
- Peu fréquent : insuffisance hépatique.
- Affections musculosquelettiques et systémiques :
- Fréquence indéterminée : myopathie et/ou rhabdomyolyse, formation de fistule, trouble de la cicatrisation des plaies, ostéonécrose de la mâchoire.
- De rares cas de myopathie et/ou de rhabdomyolyse, certains associés à une insuffisance rénale aiguë, ont été rapportés. Les patients présentant des signes ou des symptômes de toxicité musculaire devront être traités conformément à la pratique médicale courante.
- Des cas de formation de fistule ont été rapportés, parfois associés à une nécrose et une régression tumorales, et dans certains cas, à une issue fatale.
- Des cas de trouble de la cicatrisation des plaies ont été rapportés au cours du traitement par le sunitinib.
- Des cas d’ostéonécroses de la mâchoire ont été rapportés chez les patients traités par Sutent, la plupart étant apparues chez des patients ayant des facteurs de risque identifiés pour l’ostéonécrose de la mâchoire, en particulier l’exposition aux biphosphonates par voie IV et/ou un antécédent de pathologie dentaire nécessitant une intervention dentaire invasive (cf également Mises en garde et Précautions d’emploi).
- Fréquence indéterminée : myopathie et/ou rhabdomyolyse, formation de fistule, trouble de la cicatrisation des plaies, ostéonécrose de la mâchoire.
- Affections du rein et des voies urinaires :
- Fréquence indéterminée : insuffisance rénale, insuffisance rénale aiguë, protéinurie, syndrome néphrotique.
- Des cas d’altération de la fonction rénale, d’insuffisance rénale et/ou d’insuffisance rénale aiguë, dont certains ont été d’issue fatale, ont été rapportés.
- Des cas de protéinurie et de rares cas de syndrome néphrotique ont été rapportés. La poursuite du traitement par Sutent chez les patients présentant une protéinurie modérée à sévère n’a pas été évaluée systématiquement. Le traitement par Sutent doit être arrêté chez les patients qui présentent un syndrome néphrotique (cf Mises en garde et Précautions d’emploi également).
- Fréquence indéterminée : insuffisance rénale, insuffisance rénale aiguë, protéinurie, syndrome néphrotique.
- Affections pulmonaires :
- Fréquence indéterminée : épanchement pleural, embolie pulmonaire et insuffisance respiratoire.
- Des cas d’embolie pulmonaire, dont certains d’issue fatale, ont été rapportés.
- Fréquence indéterminée : épanchement pleural, embolie pulmonaire et insuffisance respiratoire.
- Investigations :
- Fréquent : augmentation de la concentration de thyrotropine (TSH).
- Fréquent : augmentation de la concentration de thyrotropine (TSH).
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : Inhibiteur de protéine kinase (code ATC : L01XE04).
- Mécanisme d’action :
- Le sunitinib inhibe plusieurs récepteurs à tyrosine kinase (RTK) impliqués dans la croissance tumorale, la néo-angiogenèse pathologique et la progression métastatique du cancer. Le sunitinib a été identifié comme un inhibiteur des récepteurs du facteur de croissance plaquettaire (PDGFRalpha et PDGFRß), des récepteurs du facteur de croissance endothélial vasculaire (VEGFR1, VEGFR2 et VEGFR3), du récepteur du facteur de cellule souche (KIT), du récepteur Fms-like tyrosine kinase-3 (FLT3), du récepteur du facteur stimulant la formation de colonies (CSF-1R) et du récepteur du facteur neurotrophique de la lignée gliale (RET). Les tests biochimiques et cellulaires ont montré que le principal métabolite du sunitinib présentait le même pouvoir inhibiteur que le sunitinib.
- Efficacité et tolérance cliniques :
- La tolérance et l’efficacité cliniques de Sutent ont été étudiées lors du traitement de patients présentant un GIST résistant à l’imatinib (patients dont la tumeur a progressé pendant ou après le traitement par l’imatinib) ou n’ayant pas toléré ce médicament (patients ayant présenté une toxicité significative pendant le traitement par l’imatinib, ayant empêché sa continuation), chez les patients atteints d’un MRCC et chez les patients atteints de pNET non résécables.
- Les données d’efficacité reposent sur le temps jusqu’à progression tumorale et sur l’allongement de la durée de survie pour les patients atteints de GIST, sur la survie sans progression pour les patients atteints de MRCC non prétraités, sur les taux de réponse objective pour les patients atteints de MRCC après échec d’un traitement à base de cytokine et sur la survie sans progression pour les patients atteints de pNET.
-
- Tumeurs stromales gastro-intestinales (GIST) :
- Une première étude ouverte, en escalade de doses, a été menée chez des patients atteints de GIST après échec d’un traitement par l’imatinib (dose médiane journalière maximale : 800 mg) dû à une résistance ou à une intolérance. 97 patients ont été inclus pour recevoir différentes doses et schémas posologiques ; 55 patients ont reçu 50 mg selon le schéma posologique recommandé de 4 semaines de traitement suivi de 2 semaines sans traitement (schéma posologique 4/2).
- Dans cette étude, le temps médian jusqu’à progression de la tumeur (Time to Tumour Progression : TTP) était de 34,0 semaines (IC à 95 % : 22,0-46,0 semaines).
- Une étude randomisée de phase III, en double aveugle, contrôlée contre placebo, a été menée chez des patients atteints de GIST intolérants à un traitement par l’imatinib ou ayant présenté une progression de la maladie pendant ou après le traitement (dose médiane journalière maximale : 800 mg). 312 patients ont été randomisés (selon un ratio de 2:1) pour recevoir soit 50 mg de Sutent, soit un placebo, administré par voie orale à raison d’une prise par jour selon un schéma posologique 4/2, jusqu’à progression de la maladie ou sortie de l’étude pour une autre raison (207 patients ont reçu Sutent et 105 ont reçu un placebo). Le critère d’efficacité principal était le TTP, défini par le temps écoulé entre la randomisation et la première confirmation d’une progression tumorale.
- Au moment de l’analyse intermédiaire prédéfinie, le TTP médian sous Sutent était de 28,9 semaines (IC à 95 % : 21,3-34,1 semaines) selon l’évaluation de l’investigateur et de 27,3 semaines (IC à 95 % : 16,0-32,1 semaines) selon l’évaluation du Comité de revue indépendant, ce qui est statistiquement significativement plus long que le TTP observé sous placebo : 5,1 semaines (IC à 95 % : 4,4-10,1 semaines) selon l’évaluation de l’investigateur et 6,4 semaines (IC à 95 % : 4,4-10,0 semaines) selon l’évaluation du Comité de revue indépendant. La survie globale était statistiquement en faveur de Sutent (hazard ratio : 0,491 [IC à 95 % : 0,290-0,831]). Le risque de mortalité était deux fois plus grand pour les patients dans le bras placebo comparé à celui des patients dans le bras Sutent.
- Après l’analyse intermédiaire de l’efficacité et de la sécurité des patients, selon la recommandation du Comité de surveillance et de suivi (Data Safety Monitoring Board DSMB) indépendant, une levée d’aveugle a été réalisée et les patients du bras placebo se sont vus offrir le traitement par Sutent en ouvert. Au total, 255 patients ont reçu Sutent dans la phase de traitement en ouvert de l’étude, dont 99 patients qui étaient initialement traités par placebo. L’analyse des critères primaires et secondaires dans la partie en ouvert de l’étude a reconfirmé les résultats obtenus au moment de l’analyse intermédiaire, tel que montré dans le tableau suivant :
-
Tableau 4 : Résumé des critères d’efficacité (population ITT) Critère Traitement en double-aveugle(a) Groupe initialement traité par placebo(b) Médiane (IC à 95 %) Hazard Ratio (HR) Sutent Placebo (IC à 95 %) p Primaire : TTP (en semaines) – Intermédiaire
27,3 (16,0 à 32,1) 6,4 (4,4 à 10,0) 0,329 (0,233 à 0,466) < 0,001 – – Final
26,6 (16,0 à 32,1) 6,4 (4,4 à 10,0) 0,339 (0,244 à 0,472) < 0,001 10,4 (4,3 à 22,0) Secondaires : PFS (en semaines)(c) – Intermédiaire
24,1 (11,1 à 28,3) 6,0 (4,4 à 9,9) 0,333 (0,238 à 0,467) < 0,001 – – Final
22,9 (10,9 à 28,0) 6,0 (4,4 à 9,7) 0,347 (0,253 à 0,475) < 0,001 – ORR (en %)(d) – Intermédiaire
6,8 (3,7 à 11,1) 0 (-) NA 0,006 – – Final
6,6 (3,8 à 10,5) 0 (-) NA 0,004 10,1 (5,0 à 17,8) OS (en semaines)(e) – Intermédiaire
– – 0,491 (0,290 à 0,831) 0,007 – – Final
72,7 (61,3 à 83,0) 64,9 (45,7 à 96,0) 0,876 (0,679 à 1,129) 0,306 – -
(a)
Les résultats du traitement en double-aveugle sont donnés pour la population ITT et en utilisant les mesures du radiologue centralisé, si approprié.
-
(b)
Résultats d’efficacité pour les 99 sujets qui ont changé de traitement du placebo vers Sutent après la levée d’aveugle. L’état initial a été repris au moment du changement de traitement et les analyses d’efficacité sont basées sur les évaluations des investigateurs.
-
(c)
Les chiffres intermédiaires de PFS ont été mis à jour suite à un recalcul des données d’origine.
-
(d)
Les résultats pour l’ORR sont donnés en pourcentages de sujets ayant montré une réponse confirmée avec un IC à 95 %.
-
(e)
La médiane n’est pas atteinte car les données ne sont pas encore matures.
- La survie globale médiane (OS : Overall Survival) dans la population ITT était de 72,7 semaines et 69,4 semaines (HR : 0,876 ; IC à 95 % : 0,679-1,129 ; p = 0,306), dans les bras Sutent et placebo respectivement. Dans cette analyse, le bras placebo incluait les patients randomisés au placebo qui ont reçu par la suite un traitement par Sutent en ouvert.
-
- Cancer du rein métastatique (MRCC) non prétraité :
- Une étude randomisée de phase III, multicentrique, internationale évaluant l’efficacité et la tolérance du sunitinib versus interféron IFN-alpha chez des patients atteints d’un MRCC non prétraités a été menée. 750 patients ont été randomisés en deux groupes de traitement selon un ratio de 1/1, à savoir un groupe recevant du sunitinib par cycles consécutifs de 6 semaines consistant en l’administration de 50 mg par jour de sunitinib par voie orale pendant 4 semaines, suivie de 2 semaines de fenêtre thérapeutique (schéma 4/2), et un groupe recevant l’IFN-alpha en injection sous-cutanée de 3 millions d’unités (MU) la 1re semaine, 6 MU la 2e semaine et 9 MU la 3e semaine et ensuite, chaque semaine suivante, 3 injections effectuées à des jours non consécutifs.
- La durée médiane de traitement était de 11,1 mois (durée allant de 0,4 à 46,1 mois) pour le traitement par le sunitinib et de 4,1 mois (durée allant de 0,1 à 45,6 mois) pour le traitement par l’IFN-alpha. Des événements indésirables graves liés au traitement ont été rapportés chez 23,7 % des patients recevant du sunitinib et chez 6,9 % des patients recevant l’IFN-alpha. Cependant, les taux d’arrêt du traitement liés aux événements indésirables étaient de 20 % pour le sunitinib et de 23 % pour l’IFN-alpha. Les interruptions de traitement ont eu lieu chez 202 patients (54 %) traités par le sunitinib et 141 patients (39 %) traités par l’IFN-alpha. Des réductions de doses ont eu lieu chez 194 patients (52 %) traités par le sunitinib et chez 98 patients (27%) traités par l’IFN-alpha. Les patients ont été traités jusqu’à progression de la maladie ou jusqu’à la sortie de l’étude.
- Le critère principal d’efficacité était la survie sans progression (PFS : Progression Free Survival). Une analyse intermédiaire prévue dans l’étude a montré un avantage statistiquement significatif pour Sutent comparé à l’IFN-alpha ; dans cette étude, la valeur médiane de la PFS était de 47,3 semaines dans le groupe traité par le sunitinib et de 22,0 semaines dans le groupe traité par l’IFN-alpha ; le risque relatif était égal à 0,415 (IC à 95 % : 0,320-0,539 ; valeur de p < 0,001). Les autres critères étudiés regroupaient le taux de réponse objective (ORR : Objective Response Rate), la survie globale (OS : Overall Survival) et la tolérance. L’évaluation radiologique principale a été abandonnée après l’atteinte du critère principal. Lors de l’analyse finale, le taux de réponse objective tel que déterminé par les investigateurs était de 46 % (IC à 95 % : 41-51) pour le bras sunitinib et de 12,0 % (IC à 95 % : 9-16) pour le bras IFN-alpha (p < 0,001).
- Le traitement par le sunitinib a été associé à une durée de survie plus longue comparée à l’IFN-alpha. La survie globale médiane était de 114,6 semaines pour le bras sunitinib (IC à 95 % : 100,1-142,9 semaines) et de 94,9 semaines pour le bras IFN-alpha (IC à 95 % : 77,7-117,0 semaines) avec un hazard ratio de 0,821 (IC à 95 % : 0,673-1,001 ; p = 0,0510 par le test du log-rank non stratifié).
- La survie sans progression et la survie globale, observées dans la population ITT, et telles que déterminées par l’évaluation clinique radiologique principale, sont résumées dans le tableau ci-dessous :
-
Résumé des critères d’efficacité (population ITT) Sunitinib (N = 375) IFN-alpha (N = 375) Résumé de la survie sans progression Sujets qui n’ont pas progressé ou qui ne sont pas décédés (n [%]) 161 (42,9) 176 (46,9) Sujets identifiés comme ayant progressé ou étant décédés (n [%]) 214 (57,1) 199 (53,1) PFS (en semaines) Quartile (IC à 95 %) 25 % 22,7 (18,0 à 34,0) 10,0 (7,3 à 10,3) 50 % 48,3 (46,4 à 58,3) 22,1 (17,1 à 24,0) 75 % 84,3 (72,9 à 95,1) 58,1 (45,6 à 82,1) Analyse non stratifiée Hazard Ratio (sunitinib vs IFN-alpha) 0,5268 IC à 95 % pour le Hazard Ratio (0,4316 à 0,6430) Valeur de p(a) < 0,0001 Résumé de la survie globale Sujets non identifiés comme décédés (n [%]) 185 (49,3) 175 (46,7) Sujets identifiés décédés (n [%]) 190 (50,7) 200 (53,3) Survie globale (en semaines) Quartile (IC à 95 %) 25 % 56,6 (48,7 à 68,4) 41,7 (32,6 à 51,6) 50 % 114,6 (100,1 à 142,9) 94,9 (77,7 à 117,0) 75 % NA(b) (NA à NA) NA(b) (NA à NA) Analyse non stratifiée Hazard Ratio (sunitinib vs IFN-alpha) 0,8209 IC à 95 % pour le Hazard Ratio (0,6730 à 1,0013) Valeur de p(a) 0,0510 -
(a)
à partir du test du log-rank bilatéral
-
(b)
NA : non applicable (non atteint)
-
- Cancer du rein métastatique (MRCC) après échec d’un traitement par cytokine :
- Une étude de phase II a été conduite chez des patients après échec d’un traitement préalable par cytokine à base d’interleukine-2 ou d’IFN-alpha. 63 patients ont reçu 50 mg de sunitinib par voie orale, à raison d’une prise par jour pendant 4 semaines consécutives, suivies d’une fenêtre thérapeutique de 2 semaines, correspondant à un cycle complet de 6 semaines (schéma posologique 4/2). Le critère d’efficacité principal était le taux de réponse objective (ORR) calculé d’après les critères d’évaluation des réponses tumorales relatives aux tumeurs solides (RECIST).
- Dans cette étude, le taux de réponse objective était de 36,5 % (IC à 95 % : 24,7-49,6 %) et le temps médian jusqu’à la progression (TTP) de 37,7 semaines (IC à 95 % : 24,0-46,4 semaines).
- Une étude confirmatoire multicentrique, en ouvert, non contrôlée, évaluant l’efficacité et la tolérance de Sutent, a été menée chez des patients présentant un cancer du rein métastatique après échec d’un précédent traitement par cytokine. 106 patients ont reçu au moins une dose de 50 mg de Sutent selon le schéma 4/2.
- Le principal critère d’efficacité de cette étude était le taux de réponse objective (ORR). Les critères d’évaluation secondaires comprenaient le TTP, la durée de la réponse (DR) et la survie globale (OS).
- Dans cette étude, le taux ORR était de 35,8 % (IC à 95 % : 26,8-47,5 %) ; les valeurs médianes de la durée de la réponse et de la survie globale n’avaient pas encore été atteintes.
-
- Tumeurs neuroendocrines du pancréas (pNET) :
- Une étude support de phase II, ouverte, multicentrique, a évalué l’efficacité et la tolérance de Sutent en monothérapie à une dose journalière de 50 mg selon le schéma 4/2 [4 semaines de traitement, 2 semaines de fenêtre thérapeutique] chez des patients atteints de pNET non résécables. Dans une cohorte de 66 patients atteints d’une tumeur des îlots de Langerhans, le taux de réponse, critère principal, était de 17 %.
- Une étude pivot de phase III, multicentrique, internationale, randomisée, en double aveugle, portant sur le sunitinib en monothérapie, le groupe contrôle étant sous placebo, a été menée chez des patients atteints de pNET non résécables.
- Les patients devaient avoir présenté une progression documentée, sur la base des critères RECIST, dans les 12 derniers mois et ont été randomisés (1:1) pour recevoir soit une dose journalière de 37,5 mg de sunitinib sans fenêtre thérapeutique préétablie (n = 86) soit le placebo (n = 85).
- L’objectif principal était de comparer la survie sans progression (PFS) des patients recevant le sunitinib versus les patients recevant le placebo. D’autres critères comprenaient la survie globale (OS), le taux de réponse objective (ORR), les résultats de l’auto-évaluation des patients (PRO) et la tolérance.
- Les données démographiques étaient comparables entre le groupe sunitinib et le groupe placebo. De plus, 49 % des patients recevant le sunitinib avaient une tumeur non fonctionnelle comparativement à 52 % des patients recevant le placebo, et 92 % des patients dans les deux bras présentaient des métastases hépatiques.
- L’utilisation d’analogues de la somatostatine a été autorisée dans l’étude.
- Un total de 66 % des patients sous sunitinib avaient reçu une thérapie par voie systémique antérieure comparativement à 72 % des patients sous placebo. De plus, 24 % des patients du groupe sunitinib avaient reçu des analogues de la somatostatine contre 22 % des patients du groupe placebo.
- Un avantage cliniquement significatif pour la PFS évaluée par les investigateurs a été observé avec le sunitinib par rapport au placebo. La PFS médiane était de 11,4 mois pour le bras sunitinib comparé à 5,5 mois pour le bras placebo [hazard Ratio : 0,418 (IC à 95 % : 0,263-0,662), valeur de p = 0,0001]. Des résultats similaires ont été observés lorsque l’évaluation de la réponse tumorale basée sur l’application des critères RECIST pour des mesures tumorales faites par l’investigateur était utilisée pour déterminer la progression de la maladie, cf tableau 5.
- Un hazard ratio (risque relatif) en faveur de Sutent a été observé dans tous les sous-groupes de patients, déterminés sur leurs caractéristiques à l’entrée dans l’étude, y compris une analyse par nombre de traitements systémiques antérieurs. Un total de 29 patients dans le bras sunitinib et 24 dans le bras placebo n’avaient reçu aucun traitement systémique antérieur ; parmi ces patients, le risque relatif pour la PFS était de 0,365 (IC à 95 % : 0,156-0,857), p = 0,0156. De la même manière, parmi les 57 patients dans le bras sunitinib (dont 28 ayant reçu 1 thérapie systémique antérieure et 29 ayant reçu 2 thérapies systémiques antérieures ou plus), et parmi les 61 patients dans le bras placebo (dont 25 patients ayant reçu 1 thérapie systémique antérieure et 36 ayant reçu 2 thérapies systémiques antérieures ou plus), qui avaient tous reçu au moins une thérapie systémique antérieure, le risque relatif pour la PFS était de 0,456 (IC à 95 % : 0,264-0,787), p = 0,0036.
- Une analyse de sensibilité de la PFS a été menée au cours de laquelle la progression était basée sur les mesures tumorales rapportées par l’investigateur et au cours de laquelle tous les patients censurés pour toute raison autre que l’arrêt de l’étude, ont été considérés comme ayant présenté un événement de PFS. Cette analyse a donné une estimation prudente de l’effet du traitement par le sunitinib et a conforté l’analyse primaire, en démontrant un risque relatif de 0,507 (IC à 95 % : 0,350-0,733), p = 0,000193. L’étude pivot dans les tumeurs neuroendocrines du pancréas a été terminée prématurément selon les recommandations du Comité de surveillance (Drug Monitoring Committee) indépendant, et le critère principal a été basé sur l’évaluation de l’investigateur, ce qui a pu impacter les estimations de l’effet du traitement.
- Afin d’éliminer un biais dans l’évaluation de la PFS par l’investigateur, une revue en aveugle, centralisée et indépendante des scanners a été réalisée et a confirmé l’évaluation de la PFS par l’investigateur, cf tableau 5.
-
Tableau 5 : Résultats d’efficacité de l’étude de phase III pNET Paramètres d’efficacité Sutent (n = 86) Placebo (n = 85) HR (IC à 95 %) Valeur de p Évaluation de la survie sans progression [médiane, mois (IC à 95 %)] par l’investigateur 11,4 (7,4-19,8) 5,5 (3,6-7,4) 0,418 (0,263-0,662) 0,0001(a) Survie sans progression [médiane, mois (IC à 95 %)] par l’évaluation de la réponse tumorale basée sur l’application des critères RECIST à l’évaluation tumorale par l’investigateur 12,6 (7,4-16,9) 5,4 (3,5-6,0) 0,401 (0,252-0,640) 0,000066(a) Survie sans progression [médiane, mois (IC à 95 %)] par la revue en aveugle, centralisée et indépendante de l’évaluation de la tumeur 12,6 (11,1-20,6) 5,8 (3,8-7,2) 0,315 (0,181-0,546) 0,000015(a) Survie globale [médiane, mois (IC à 95 %)] 20,6 (20,6-NR) NR (15,5-NR) 0,409 (0,187-0,894) 0,0204(a) Taux de réponse objective 9,3 (3,2-15,4) 0 NA 0,0066(b) - IC = intervalle de confiance, HR = hazard ratio, NA = non applicable, NR = non atteint
-
(a)
test du log-rank bilatéral non stratifié
-
(b)
test exact de Fisher
-
Figure 1 : Courbe de la PFS de Kaplan-Meier dans l’étude de phase III pNET 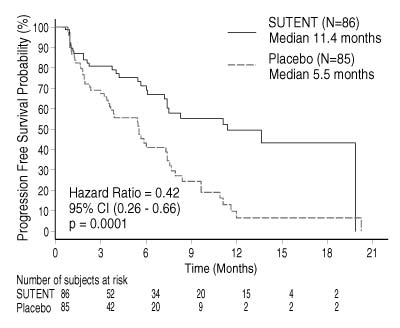
Abscisse = temps (mois), ordonnée = probabilité de survie sans progression (%) ; Sutent (N = 86), médiane 11,4 mois ; Placebo (N = 85), médiane 5,5 mois ; Hazard Ratio = 0,42, IC à 95 % (0,26-0,66), p = 0,0001 ; Nombre de sujets à risque.
- Les données de survie globale n’étaient pas matures au moment de l’analyse. Il y a eu 9 décès dans le bras sunitinib et 21 décès dans le bras placebo. Une différence statistiquement significative en faveur du sunitinib par rapport au placebo a été observée sur le taux de réponse objective.
- En cas de progression de la maladie, une levée d’aveugle était effectuée et les patients sous placebo ont eu la possibilité de prendre Sutent en ouvert dans une étude d’extension séparée. En conséquence de l’arrêt prématuré de l’étude, une levée d’aveugle a été effectuée pour les patients restants dans celle-ci et ils ont eu la possibilité de prendre Sutent en ouvert dans une étude d’extension. Un total de 59 patients du bras placebo ont reçu Sutent dans une étude d’extension.
- Les résultats du questionnaire de l’organisation européenne pour la recherche et le traitement du cancer et de la qualité de vie (EORTC QLQC-30) ont démontré que la qualité de vie globale liée à la santé et les cinq domaines fonctionnels (physique, rôle, cognitif, émotionnel et social) ont été maintenus pour les patients recevant le sunitinib en comparaison avec le placebo avec des effets indésirables symptomatiques limités.
-
- Population pédiatrique :
- L’Agence européenne du médicament (EMA) a différé l’obligation de soumettre les résultats d’études réalisées avec Sutent dans un ou plusieurs sous-groupes de la population pédiatrique dans les tumeurs stromales gastro-intestinales (GIST) : pour les informations concernant l’usage pédiatrique, cf Posologie et Mode d’administration.
- L’EMA a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec Sutent dans tous les sous-groupes de la population pédiatrique dans le traitement du carcinome rénal et du bassinet (à l’exclusion du néphroblastome, du néphroblastomatose, du sarcome à cellule claire, du néphrome mésoblastique, du carcinome médullaire rénal et de la tumeur rhabdoïde du rein) : pour les informations concernant l’usage pédiatrique, cf Posologie et Mode d’administration.
- L’EMA a accordé une dérogation à l’obligation de soumettre les résultats des études avec Sutent dans tous les sous-groupes de la population pédiatrique dans le traitement des tumeurs neuroendocrines gastro-entéro-pancréatiques (à l’exclusion des neuroblastomes, neuroganglioblastomes et phéochromocytome) : pour les informations concernant l’usage pédiatrique, cf Posologie et Mode d’administration.
PHARMACOCINÉTIQUE |
La pharmacocinétique du sunitinib a été étudiée chez 135 volontaires sains et 266 patients présentant une tumeur solide. Les données pharmacocinétiques étaient similaires dans toutes les populations de patients présentant des tumeurs solides et chez les volontaires sains.
Dans l’éventail de doses comprises entre 25 et 100 mg, l’aire sous la courbe de concentration plasmatique en fonction du temps (AUC) et la Cmax augmentent de façon proportionnelle en fonction de la dose. Sous l’effet répété des doses journalières, le sunitinib s’accumule et sa concentration est multipliée par 3 à 4, et celle de son principal métabolite actif par 7 à 10. Les concentrations à l’équilibre du sunitinib et de son principal métabolite actif sont atteintes en 10 à 14 jours. Au 14e jour, les concentrations plasmatiques cumulées du sunitinib et de son métabolite actif sont comprises entre 62,9 et 101 ng/ml et correspondent aux concentrations cibles, anticipées d’après les résultats des études précliniques, qui inhibent in vitro la phosphorylation du récepteur et provoquent l’arrêt ou le ralentissement de la croissance tumorale in vivo. Le principal métabolite actif représente 23 à 37 % de l’exposition au médicament. Aucune modification significative de la pharmacocinétique du sunitinib ou de son principal métabolite actif n’a été observée lors de l’administration de doses journalières répétées, ni lors de cycles répétés avec les schémas posologiques testés.
- Absorption :
- Les concentrations maximales (Cmax) de sunitinib sont généralement atteintes entre 6 à 12 heures (Tmax) après l’administration orale de sunitinib.
- La biodisponibilité du sunitinib n’est pas affectée par la prise de nourriture.
- Distribution :
- In vitro, la liaison du sunitinib et de son principal métabolite actif aux protéines plasmatiques humaines, est respectivement de 95 et 90 % et ne semble pas dépendre de la concentration. Le volume de distribution apparent du sunitinib (Vd) est important (2230 l), ce qui indique une distribution tissulaire.
- Interactions métaboliques :
- Les valeurs de Ki calculées in vitro pour toutes les isoformes du cytochrome CYP testées (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5 et 4A9/11) indiquent qu’il est peu probable que le sunitinib et son principal métabolite actif induisent le métabolisme d’autres substances actives susceptibles d’être métabolisés par ces enzymes, de manière cliniquement significative.
- Biotransformation :
- Le sunitinib est principalement métabolisé par le CYP3A4, isoforme du cytochrome P450, qui produit son principal métabolite actif le déséthyl de sunitinib, lequel est ensuite de nouveau métabolisé par la même isoenzyme.
- L’administration concomitante de Sutent et d’inducteurs puissants ou d’inhibiteurs du CYP3A4 doit être évitée en raison d’une altération possible des niveaux plasmatiques de sunitinib (cf Mises en garde et Précautions d’emploi, Interactions).
- Élimination :
- L’excrétion se fait principalement par les selles (61 %) ; seulement 16 % de la dose de sunitinib administrée est éliminée par voie rénale sous la forme de substance active inchangée ou de ses métabolites. Le sunitinib et son principal métabolite actif représentent la plus grande partie des composés retrouvés dans le plasma, l’urine et les selles, soit respectivement 91,5 %, 86,4 % et 73,8 % de la radioactivité mesurée sur des échantillons groupés. Des métabolites mineurs ont été identifiés dans les urines et les selles, mais n’ont généralement pas été retrouvés dans le plasma. Les valeurs de la clairance orale totale (CL/F) étaient comprises entre 34 et 62 l/h. Après administration orale chez des volontaires sains, les demi-vies d’élimination du sunitinib et de son principal métabolite actif déséthylé sont respectivement comprises entre environ 40 et 60, et 80 et 110 heures.
- Populations particulières :
-
- Insuffisance hépatique :
Le sunitinib et son métabolite primaire sont essentiellement métabolisés par le foie. Chez les patients atteints d’une insuffisance hépatique légère ou modérée (classes A et B de Child-Pugh) et chez les patients ayant une fonction hépatique normale, les concentrations systémiques étaient similaires, après l’administration d’une dose unique de sunitinib. Sutent n’a pas été étudié chez les patients atteints d’une insuffisance hépatique sévère (classe C de Child-Pugh). - Les patients, dont les taux d’ALAT ou d’ASAT étaient supérieurs à 2,5 x LSN (limite supérieure de la normale) ou supérieurs à 5,0 x LSN en présence de métastases hépatiques, ont été exclus des études chez les patients atteints d’un cancer.
- Insuffisance rénale : Les analyses de pharmacocinétique de population ont indiqué que la clairance apparente de sunitinib (CL/F) n’était pas affectée par la clairance de la créatinine dans les fourchettes de valeurs évaluées (42-347 ml/min). Chez les patients présentant une altération sévère de la fonction rénale (CLcr < 30 ml/min) et chez les patients ayant une fonction rénale normale (CLcr > 80 ml/min), les concentrations plasmatiques étaient similaires après l’administration d’une dose unique de Sutent. Bien que le sunitinib et son métabolite principal ne soient pas éliminés par hémodialyse chez les patients présentant une IRT, les concentrations plasmatiques totales étaient inférieures de 47 % pour le sunitinib et de 31 % pour son métabolite principal par rapport aux patients ayant une fonction rénale normale.
- Poids, indice de performance : Les analyses pharmacocinétiques de population de données démographiques indiquent qu’aucun ajustement de la dose initiale n’est nécessaire en fonction du poids, ou de l’indice de performance Eastern Cooperative Oncology Group (ECOG).
- Genre : Les données disponibles indiquent que les femmes sont susceptibles de présenter une clairance apparente (CL/F) du sunitinib de 30 % inférieure à celles des hommes : néanmoins cette différence ne nécessite pas de procéder à des ajustements de la dose initiale.
- Insuffisance hépatique :
SÉCURITE PRÉCLINIQUE |
Chez le rat et le singe, des études de toxicité à doses répétées sur une durée maximale de 9 mois, ont montré que les principaux organes cibles sont le tractus gastro-intestinal (vomissements et diarrhée chez le singe) ; la glande surrénale (hyperémie du cortex surrénalien et/ou une hémorragie chez le rat et le singe, associée à une nécrose suivie d’une fibrose chez le rat) ; le système hémato-lymphopoïétique (moelle hypocellulaire et déplétion lymphoïde du thymus, de la rate et des ganglions lymphatiques) ; le pancréas exocrine (dégranulation des cellules acineuses associées à une nécrose cellulaire) ; les glandes salivaires (hypertrophie acineuse) ; les articulations (épaississement du cartilage de conjugaison) ; l’utérus (atrophie) ; les ovaires (diminution du développement folliculaire). Tous ces effets ont été observés à des niveaux d’exposition plasmatique de sunitinib cliniquement pertinents. D’autres effets ont été observés dans d’autres études, et notamment un allongement de l’intervalle QTc, une réduction de la fraction d’éjection (FEVG), une hypertrophie de l’hypophyse, une atrophie tubulaire des testicules, une augmentation du volume de la matrice mésengiale du rein, des hémorragies du tractus gastro-intestinal et de la muqueuse buccale, et une hypertrophie des cellules de l’hypophyse antérieure. Les modifications de l’utérus (atrophie de l’endomètre) et des cartilages de conjugaison (épaississement ou dysplasie des cartilages) sont considérées comme liées à l’action pharmacologique du sunitinib. La plupart de ces effets ont été réversibles et ont disparu 2 à 6 semaines après l’arrêt du traitement.
- Génotoxicité :
- Le potentiel génotoxique du sunitinib a été évalué in vitro et in vivo. Le sunitinib n’est pas mutagène chez des bactéries testées avec activation métabolique en utilisant des cellules hépatiques de rats. Le sunitinib n’induit pas d’aberrations chromosomiques dans des lymphocytes périphériques humains in vitro. Une polyploïdie (anomalie du nombre de chromosomes) a été observée dans des lymphocytes périphériques humains in vitro, avec ou sans activation métabolique. Le sunitinib n’est pas clastogène sur la moelle osseuse de rat in vivo. Le principal métabolite actif n’a pas fait l’objet d’une évaluation du potentiel génotoxique.
- Carcinogénicité :
- Le potentiel de carcinogénicité du sunitinib a été évalué chez les souris transgéniques rasH2. Des carcinomes gastroduodénaux, une élévation de l’incidence des hémangiosarcomes sous-jacents et/ou une hyperplasie de la muqueuse gastrique ont été rapportés à des doses >= 25 mg/kg/jour après une durée de traitement de 1 ou 6 mois (>= 7,3 fois l’ASC chez les patients ayant reçu la dose recommandée journalière). Aucune modification de la prolifération n’a été observée chez les souris transgéniques rasH2 à la dose de 8 mg/kg/jour (>= 0,7 fois l’ASC chez les patients ayant reçu la dose recommandée journalière). La pertinence des résultats de carcinogénicité observés chez la souris transgénique rasH2 pour l’homme après un traitement par le sunitinib sur une durée de 1 ou 6 mois n’est pas claire.
- Toxicité sur la reproduction et sur le développement du foetus :
- Aucun effet sur la fertilité mâle ou femelle n’a été observé dans les études de toxicité sur la reproduction. Cependant, dans des études de toxicité à doses répétées menées chez des rats et des singes, des effets sur la fertilité femelle ont été observés, avec une atrésie folliculaire, une dégénérescence du corps jaune, des modifications de l’endomètre et une diminution du poids de l’utérus et des ovaires, à des expositions systémiques cliniquement pertinentes. Chez le rat, des effets sur la fertilité mâle ont été observés, avec une atrophie tubulaire des testicules, une réduction du nombre de spermatozoïdes dans l’épididyme, une déplétion colloïdale dans la prostate et les vésicules séminales, à des expositions plasmatiques 18 fois supérieures à celles observées en clinique.
- Chez le rat, la mortalité embryo-foetale a été mise en évidence à travers une réduction significative du nombre de foetus vivants, une augmentation du nombre de résorptions foetales, une augmentation de la perte postimplantatoire et une perte de toute la portée chez 8 des 28 femelles gravides, à des expositions plasmatiques 5,5 fois supérieures à l’exposition clinique. Chez le lapin, les diminutions de poids de l’utérus gravide et du nombre de foetus vivants étaient dues à l’augmentation du nombre de résorptions et de l’incidence des pertes postimplantatoires et à la perte de toute la portée chez 4 des 6 femelles gravides exposées à des doses 3 fois supérieures à celles de la pratique clinique.
- Le traitement des rates par le sunitinib, au cours de la période de l’organogenèse, a provoqué des effets sur le développement foetal consistant en une augmentation de l’incidence des malformations squelettiques du foetus, principalement caractérisées par un retard de l’ossification des vertèbres thoraciques et/ou lombaires, survenus chez le rat à des expositions plasmatiques 6 fois supérieures à l’exposition clinique. Chez le lapin, ces effets consistaient en une augmentation de l’incidence des fentes labiales à des expositions plasmatiques environ équivalentes à celles utilisées en pratique clinique, et en une augmentation des fentes labiales et palatines à des expositions 2,7 fois supérieures à l’exposition clinique.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
Pas de précautions particulières de conservation.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament soumis à prescription hospitalière. Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie. Médicament nécessitant une surveillance particulière pendant le traitement. | |
| AMM | EU/1/06/347/004 ; CIP 3400938210224 (RCP rév 20.12.2010) 12,5 mg. |
| EU/1/06/347/005 ; CIP 3400938210392 (RCP rév 20.12.2010) 25 mg. | |
| EU/1/06/347/006 ; CIP 3400938210453 (RCP rév 20.12.2010) 50 mg. | |
| Prix : | 1438.05 euros (28 gélules à 12,5 mg). |
| 2850.45 euros (28 gélules à 25 mg). | |
| 5675.25 euros (28 gélules à 50 mg). | |
|
Remb Séc soc à 100 %. Non remboursable à la date du 07.01.2011 dans l’indication « Tumeurs neuroendocrines du pancréas non résécables ou métastatiques, bien différenciées, avec progression de la maladie chez l’adulte ». Collect. |
|
Pfizer Ltd, Ramsgate Road, Sandwich, Kent CT13 9NJ, Royaume-Uni.
PFIZER
23-25, av du Dr-Lannelongue. 75014 Paris
Tél : 01 58 07 30 00
Info médic : Tél : 01 58 07 34 40
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


