panitumumab
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p flacon | ||
| de 5 ml | de 20 ml | |
| Panitumumab (DCI)* | 100 mg | 400 mg |
Teneur en sodium : 0,150 mmol/ml, soit 3,45 mg/ml.
Conformément aux instructions de la rubrique Modalités de manipulation/Élimination, la concentration finale de panitumumab après préparation ne devra pas dépasser 10 mg/ml.
* Le panitumumab est un anticorps monoclonal IgG2 humain, produit à partir d’une lignée cellulaire de mammifère (CHO) par la technique de l’ADN recombinant.
INDICATIONS |
POSOLOGIE ET MODE D’ADMINISTRATION |
Le traitement par Vectibix doit être contrôlé par un médecin ayant l’expérience des traitements anticancéreux.
La détection de l’expression du gène KRAS non muté doit être réalisée par un laboratoire expérimenté utilisant un test validé.
La dose recommandée de Vectibix est de 6 mg/kg de poids corporel administrée une fois toutes les deux semaines. Avant la perfusion, Vectibix doit être dilué dans une solution injectable de chlorure de sodium à 0,9 %, la concentration finale ne devant pas dépasser 10 mg/ml (pour les instructions de préparation, cf Modalités de manipulation et d’élimination).
- Populations particulières :
- La tolérance et l’efficacité de Vectibix n’ont pas été étudiées chez les patients atteints d’insuffisance rénale ou hépatique.
- Il n’est pas nécessaire de procéder à une adaptation posologique chez les personnes âgées. Lors des essais cliniques, aucune différence globale en terme de tolérance ou d’efficacité n’a été observée entre les patients de plus de 65 ans et les patients plus jeunes.
- Aucune donnée n’étant disponible chez l’enfant, Vectibix ne doit pas être utilisé chez les patients de moins de 18 ans.
Mode d’administration :
Vectibix doit être administré par perfusion intraveineuse (IV) à l’aide d’une pompe à perfusion, en utilisant un filtre en ligne à faible pouvoir de fixation protéique de 0,2 ou 0,22 micron, par une voie d’abord périphérique ou un cathéter tunnelisé. La durée de perfusion recommandée est de 60 minutes environ. Les doses supérieures à 1000 mg doivent être administrées pendant une durée approximative de 90 minutes (pour les instructions de manipulation, cf Modalités de manipulation et d’élimination).
Le cathéter doit être rincé avec une solution de chlorure de sodium avant et après l’administration de Vectibix afin d’éviter toute interaction avec d’autres médicaments ou d’autres solutions intraveineuses.
Ne pas administrer par voie IV directe ou en bolus.
Pour les instructions de dilution du produit avant administration, cf Modalités de manipulation et d’élimination.
CONTRE-INDICATIONS |
- Patients ayant un antécédent d’hypersensibilité, sévère ou mettant en jeu le pronostic vital, à la substance active ou à l’un des excipients (cf Mises en garde et Précautions d’emploi).
- Patients présentant une pneumopathie interstitielle ou une fibrose pulmonaire (cf Mises en garde et Précautions d’emploi).
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- Réactions dermatologiques :
- Presque tous les patients (environ 90 %) traités par Vectibix ont présenté des réactions dermatologiques reliées au produit, effet pharmacologique observé avec les inhibiteurs des récepteurs au facteur de croissance épidermique (EGFR). La majorité de ces réactions sont d’intensité légère à modérée (cf Effets indésirables). Si un patient présente des réactions dermatologiques de grade 3 ou plus (selon les critères du NCI-CTC/CTCAE), ou si celles-ci sont jugées intolérables, l’administration de Vectibix devra être suspendue temporairement jusqu’à l’atténuation des réactions (<= au grade 2). Après avoir obtenu une amélioration <= au grade 2, réinstaurer l’administration de Vectibix en diminuant la dose initiale de moitié. Si les réactions ne réapparaissent pas, la dose de Vectibix devra être augmentée par paliers de 25 %, jusqu’à l’obtention de la dose recommandée. Si les réactions ne s’atténuent pas (<= au grade 2) après l’arrêt d’une ou deux doses de Vectibix, ou si les réactions réapparaissent ou deviennent intolérables avec 50 % de la dose initiale, l’utilisation du Vectibix devra être définitivement arrêtée.
- Lors des essais cliniques, suite à l’apparition de réactions dermatologiques sévères (dont la stomatite), des complications infectieuses (dont la septicémie), fatales dans de rares cas, ainsi que des abcès locaux nécessitant des incisions et un drainage ont été rapportés. L’apparition de complications infectieuses ou inflammatoires (dont la cellulite infectieuse) doit être surveillée chez les patients manifestant des réactions dermatologiques sévères ou présentant une aggravation de ces réactions pendant le traitement par Vectibix et un traitement adapté doit être mis en place rapidement. Il est recommandé aux patients de mettre de la crème solaire, de porter un chapeau et de limiter l’exposition au soleil pendant la durée du traitement par Vectibix et lors de l’apparition de rash/toxicités dermatologiques, la lumière du soleil pouvant exacerber toutes les réactions cutanées possibles.
- Complications pulmonaires :
- Les patients ayant des antécédents, ou présentant des signes de pneumopathie interstitielle ou de fibrose pulmonaire, ont été exclus des essais cliniques. Des cas de pneumonie interstitielle ayant été observés avec les inhibiteurs de l’EGFR, le traitement par Vectibix doit être interrompu en cas d’apparition brutale ou d’aggravation de symptômes pulmonaires, et une exploration de ces symptômes doit être effectuée au plus vite. Si une pneumopathie inflammatoire ou des infiltrats pulmonaires sont diagnostiqués, Vectibix devra être arrêté et le patient traité de façon appropriée.
- Troubles électrolytiques :
- Chez certains patients, une diminution progressive des concentrations sériques de magnésium conduisant à des hypomagnésémies sévères (grade 4) a été observée. Les patients doivent être surveillés périodiquement pour l’hypomagnésémie et l’hypocalcémie associée, préalablement à la mise en place du traitement par Vectibix, puis périodiquement jusqu’à 8 semaines après la fin du traitement (cf Effets indésirables). Le cas échéant, une supplémentation en magnésium est recommandée.
- D’autres troubles électrolytiques, incluant l’hypokaliémie, ont également été observés. Le cas échéant, une supplémentation par ces électrolytes est également recommandée.
- Réactions liées à la perfusion :
- Lors d’une étude clinique, 4 % des patients ont présenté des réactions liées à la perfusion, et dans 1 % des cas, ces réactions ont été considérées comme sévères (NCI-CTC grades 3 et 4).
- Sur l’ensemble des études cliniques, les réactions liées à la perfusion (survenant dans les 24 heures suivant la perfusion), ont été signalées chez 3 % des patients traités par Vectibix, dont moins de 1 % ont été sévères (NCI-CTC grades 3 et 4). Après commercialisation, des réactions graves liées à la perfusion ont été rapportées, incluant de rares cas d’issue fatale. Si une réaction sévère ou mettant en jeu le pronostic vital survient pendant une perfusion ou à tout moment après une perfusion (par exemple : apparition de bronchospasmes, oedème de Quincke, hypotension, besoin de médication parentérale ou anaphylaxie), Vectibix doit être définitivement arrêté (cf Contre-indications, Effets indésirables).
- Il est nécessaire de réduire le débit de perfusion chez les patients ayant une réaction liée à la perfusion légère ou modérée (NCI-CTC grades 1 et 2) pendant toute la durée de cette perfusion. Il est recommandé de maintenir ce débit de perfusion diminué pour toutes les perfusions suivantes.
- Des réactions d’hypersensibilité survenues plus de 24 heures après la perfusion ont été rapportées dont un cas d’oedème de Quincke d’issue fatale survenu plus de 24 heures après la perfusion.
- Les patients doivent être informés de la possibilité d’apparition tardive d’une réaction d’hypersensibilité et de la nécessité de contacter leur médecin si des symptômes d’une réaction d’hypersensibilité apparaissent.
- Autres précautions :
- Prendre en compte la teneur en sodium (cf Composition), chez les patients ayant un régime sodique contrôlé.
- Vectibix en association avec le protocole IFL :
- Les patients recevant Vectibix en association avec le protocole IFL (5-fluorouracile [500 mg/m2], leucovorine [20 mg/m2] et irinotécan [125 mg/m2] en bolus) ont présenté une incidence élevée de diarrhées sévères (cf Effets indésirables). Par conséquent, l’administration de Vectibix en association avec le protocole IFL doit être évitée (cf Interactions).
- Vectibix en association avec le bévacizumab et des protocoles de chimiothérapie :
- Un essai multicentrique, randomisé, en ouvert, mené chez 1053 patients, a permis d’évaluer l’efficacité du bévacizumab et des protocoles de chimiothérapie à base d’oxaliplatine ou d’irinotécan associés ou non au Vectibix dans le traitement du cancer colorectal métastatique en première ligne. Lors de l’analyse intermédiaire conduite chez 947 patients randomisés, une diminution de la durée de la survie sans progression et une augmentation de l’incidence de décès chez les patients recevant Vectibix associé au bévacizumab et à la chimiothérapie ont été observées. Une plus grande incidence d’embolie pulmonaire, d’infections (d’origine principalement dermatologique), de diarrhées, de troubles électrolytiques et de déshydratation a également été observée dans le bras de traitement utilisant Vectibix en association avec le bévacizumab et la chimiothérapie. Une analyse complémentaire des données d’efficacité en fonction du statut KRAS n’a pas révélé de sous-groupe de patients bénéficiant de l’association de Vectibix aux chimiothérapies à base d’oxaliplatine ou d’irinotécan et de bévacizumab. Une tendance à la dégradation du taux de survie a été observée avec Vectibix dans le sous-groupe de patients exprimant le KRAS type sauvage et inclus dans la cohorte oxaliplatine. Cette même tendance à la dégradation du taux de survie a été observée avec Vectibix dans la cohorte irinotécan, indépendamment du statut mutationnel KRAS. Par conséquent, Vectibix ne doit pas être administré en association avec les chimiothérapies associant du bévacizumab (cf Interactions, Pharmacodynamie).
- Vectibix en association avec une chimiothérapie à base d’oxaliplatine chez les patients atteints de cancer colorectal métastatique :
- Vectibix ne doit pas être administré en association avec une chimiothérapie à base d’oxaliplatine chez les patients atteints de CCRm présentant une mutation KRAS ou chez lesquels le statut KRAS n’a pas été déterminé. Lors d’un essai de phase 3 (n = 1183, 656 patients avec le statut KRAS sauvage et 440 patients avec le statut KRAS muté) en première ligne de traitement du CCRm évaluant le panitumumab en association avec une perfusion de 5-fluorouracile, leucovorine et oxaliplatine (FOLFOX) versus FOLFOX seul, la survie sans progression et la survie globale ont été plus courtes chez les patients KRAS muté ayant reçu le panitumumab et le FOLFOX (n = 221) vs le FOLFOX seul (n = 219).
- Insuffisance rénale aiguë :
- Une insuffisance rénale aiguë a été observée chez des patients présentant une diarrhée sévère et une déshydratation.
INTERACTIONS |
Aucune étude d’interaction médicamenteuse n’a été réalisée.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Aucune donnée n’est disponible sur l’utilisation de Vectibix chez la femme enceinte. Les études chez l’animal ont mis en évidence une toxicité sur la reproduction (cf Sécurité préclinique). Le risque potentiel n’est pas connu dans l’espèce humaine. L’EGFR est impliqué dans le contrôle du développement prénatal et pourrait s’avérer essentiel à l’organogenèse, à la prolifération et à la différenciation normales de l’embryogenèse. Par conséquent, Vectibix peut présenter un risque potentiel pour le foetus en cas d’administration chez la femme enceinte.
L’IgG humaine traversant la barrière placentaire, le panitumumab peut être transmis de la mère au foetus. Les femmes en âge de procréer doivent utiliser des mesures contraceptives efficaces au cours du traitement par Vectibix et pendant les 6 mois qui suivent la dernière administration. Si Vectibix est utilisé pendant la grossesse ou en cas de grossesse survenant au cours du traitement, la patiente devra être avertie des risques potentiels d’interruption de grossesse ou des risques potentiels pour le foetus.
Fertilité :
Des études conduites chez l’animal ont montré des effets réversibles sur le cycle menstruel et une réduction de la fertilité des singes femelles (cf Sécurité préclinique). Le panitumumab peut affecter la capacité d’une femme à être enceinte.
Allaitement :
Le passage du panitumumab dans le lait maternel n’est pas connu. L’IgG humaine étant sécrétée dans le lait maternel, le panitumumab pourrait l’être également. La possibilité d’absorption et ses conséquences pour le nourrisson après ingestion ne sont pas connues. Il est recommandé aux femmes de ne pas allaiter pendant le traitement par Vectibix et pendant 3 mois après administration de la dernière dose.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
Les effets indésirables fréquemment rapportés chez >= 20 % des patients sont des troubles gastro-intestinaux (nausées [30 %], diarrhées [27 %] et vomissements (22 %]) ; des troubles généraux (fatigue [35 %]), des infections et infestations (paronychie [21 %]) et des affections de la peau et du tissu sous-cutané (prurit [53 %], érythème [52 %], dermatite acnéiforme [51 %], rash [38 %]).
Dans chaque niveau d’incidence, les effets indésirables sont présentés par ordre décroissant de sévérité.
- Très fréquent : dermatite acnéiforme, rash, rash exfoliatif, érythème, exfoliation de la peau, prurit, sécheresse cutanée, fissures cutanées, acné.
- Fréquent : syndrome d’érythrodysestésie palmoplantaire, rash papuleux, rash pruritique, rash érythémateux, rash maculaire, rash maculopapuleux, ulcère cutané, escarre, hypertrichose, alopécie, onychoclasie, affection unguéale (onycholyse).
- Non connu : oedème de Quincke.
- Très fréquent : diarrhées, nausées, vomissements, douleur abdominale, stomatite, constipation.
- Fréquent : sécheresse buccale.
- Très fréquent : fatigue, pyrexie.
- Fréquent : réaction liée à la perfusion, inflammation muqueuse, frissons, inconfort thoracique.
- Très fréquent : paronychie.
- Fréquent : rash pustuleux, infection oculaire, infection de la paupière, cellulite.
- Fréquent : hypomagnésémie, hypocalcémie, hypokaliémie, déshydratation.
- Très fréquent : dyspnée, toux.
- Fréquent : embolie pulmonaire, épistaxis, sécheresse nasale.
- Peu fréquent : bronchospasme.
- Fréquent : céphalées, vertiges.
- Fréquent : conjonctivite, croissance des cils, larmoiement, hyperhémie oculaire, sécheresse oculaire, prurit oculaire, irritation de la paupière, irritation oculaire.
- Fréquent : hypersensibilité.
- Peu fréquent : réaction anaphylactique.
- Fréquent : tachycardie.
- Peu fréquent : cyanose.
- Fréquent : douleur dorsale.
- Peu fréquent : hypotension, hypertension, rougeur.
* Cet effet indésirable n’a pas été rapporté au cours des essais cliniques de monothérapie (n = 1052). Le niveau d’incidence est extrait des rapports de toutes les études menées avec Vectibix (n = 4593).
- Affections gastro-intestinales :
- Les diarrhées décrites ont été d’intensité légère à modérée. 2 % des patients KRAS non muté ont présenté une diarrhée sévère. Des cas d’insuffisance rénale aiguë ont été rapportés chez les patients présentant des diarrhées et une déshydratation (cf Mises en garde et Précautions d’emploi).
- Réactions liées à la perfusion :
- Dans le cadre des réactions liées à la perfusion survenant dans les 24 heures après une perfusion, des effets indésirables incluant douleur abdominale, réactions anaphylactiques, oedème de Quincke, douleur dorsale, bronchospasme, arrêt cardiorespiratoire, douleur thoracique, frissons, cyanose, dyspnée, rougeur, hypertension, hypotension, pyrexie, tachycardie et vomissements ont été rapportés dans les essais cliniques et après commercialisation. Sur l’ensemble des essais cliniques, les réactions liées à la perfusion survenant dans les 24 heures après une perfusion ont été rapportées chez 3 % des patients traités par Vectibix, parmi lesquelles < 1 % étaient sévères (NCI-CTC de grades 3 et 4). Après commercialisation, des réactions graves liées à la perfusion ont été rapportées, incluant de rares cas d’issue fatale.
- Un cas d’oedème de Quincke d’issue fatale a été rapporté chez un patient atteint d’un carcinome métastatique squameux et récurrent de la tête et du cou traité par Vectibix lors d’un essai clinique. Cet événement est survenu lors de la réintroduction du Vectibix, suite à un premier épisode d’oedème de Quincke ; ces deux événements sont survenus plus de 24 heures après l’administration (cf Contre-indications, Mises en garde et Précautions d’emploi). Des réactions d’hypersensibilité survenant plus de 24 heures après une perfusion ont aussi été rapportées après commercialisation.
- Pour la prise en charge clinique des réactions liées à la perfusion, cf Mises en garde et Précautions d’emploi.
- Affections de la peau et du tissu sous-cutané :
- L’éruption cutanée est apparue le plus fréquemment sur le visage, le haut de la poitrine et le dos, mais a pu s’étendre aux extrémités. Suite à l’apparition de réactions cutanées et sous-cutanées sévères, des complications infectieuses telles que la septicémie, fatales dans de rares cas, des cellulites infectieuses ainsi que des abcès locaux nécessitant des incisions et un drainage ont été rapportées. Le délai médian d’apparition du premier symptôme de réaction dermatologique a été de 10 jours, et le délai médian de résolution après la dernière dose de Vectibix a été de 28 jours.
- L’inflammation péri-unguéale a été associée à un gonflement des replis cutanés latéraux des orteils et des doigts.
- Les réactions dermatologiques (incluant les affections unguéales), observées chez des patients traités par Vectibix ou autres inhibiteurs de l’EGFR, sont connues pour être associées aux effets pharmacologiques du traitement. Les effets indésirables sévères (grades 3 et 4) issus des données globales des traitements du CCRm par monothérapie incluent dermatite acnéiforme (5 %), érythème (4 %), rash (3 %), prurit (2 %), rash exfoliatif (1 %), acné (1 %), fissure cutanée (1 %), exfoliation de la peau (< 1 %), peau sèche (< 1 %), ulcère cutané (< 1 %), escarre (< 1 %), rash érythémateux (< 1 %), rash papuleux (< 1 %) et rash maculopapuleux (< 1 %). Une paronychie a été observée chez 1 % des patients traités par Vectibix.
- Vectibix en association avec d’autres agents anticancéreux et/ou monothérapie :
- Sur l’ensemble des essais cliniques, en association avec d’autres agents anticancéreux et/ou monothérapie, les effets indésirables les plus graves associés au Vectibix ont été les embolies pulmonaires, les toxicités cutanées sévères compliquées par des séquelles infectieuses et décès par choc septique, les réactions liées à la perfusion et l’hypomagnésémie. Les effets indésirables ayant nécessité un arrêt du traitement par Vectibix ont été les réactions liées à la perfusion, les toxicités cutanées sévères et la paronychie.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : agents antinéoplasiques, anticorps monoclonaux (code ATC : L01XC08).
- Mécanisme d’action :
- Le panitumumab est un anticorps monoclonal IgG2 recombinant entièrement humain se liant avec une grande affinité et spécificité à l’EGFR humain. L’EGFR est une glycoprotéine transmembranaire, membre d’une sous-famille de récepteurs à tyrosine kinase de type I comprenant l’EGFR (HER1/c- ErbB-1), HER2, HER3, et HER4. L’EGFR favorise le développement cellulaire des tissus épithéliaux normaux, tels que la peau et les follicules pileux, et est exprimé sur un grand nombre de cellules tumorales.
- Le panitumumab se fixe au domaine de liaison du ligand de l’EGFR et inhibe l’autophosphorylation du récepteur induite par tous les ligands connus de l’EGFR. La fixation du panitumumab à l’EGFR a pour effet l’internalisation du récepteur, l’inhibition du développement cellulaire, l’induction d’une apoptose et la diminution de la production d’interleukine 8 et du facteur de croissance endothélial vasculaire.
- Le gène KRAS (Kirsten rat sarcoma 2 viral oncogène homologue) code pour une petite protéine liée au GTP, impliquée dans la transduction du signal. Différents stimuli, incluant celui de l’EGFR, activent la protéine K-ras qui stimule alors d’autres protéines intracellulaires entraînant ainsi la prolifération cellulaire, la survie cellulaire et l’angiogenèse.
- Les mutations constitutives du gène KRAS sont observées fréquemment dans différentes tumeurs humaines et sont impliquées à la fois dans l’oncogenèse et la progression tumorale.
- Effets pharmacodynamiques :
- Les tests in vitro et les études animales in vivo ont mis en évidence que le panitumumab inhibe le développement et la survie des cellules tumorales exprimant l’EGFR. Aucun effet antitumoral du panitumumab n’a été observé sur les xénogreffes tumorales humaines n’exprimant pas l’EGFR. L’ajout du panitumumab à une radiothérapie, une chimiothérapie et/ou à d’autres agents de thérapie ciblée, dans les études animales, a entraîné une augmentation des effets antitumoraux par rapport à la chimiothérapie ou aux agents thérapeutiques ciblés utilisés seuls.
- Immunogénicité :
- Les données sur l’apparition d’anticorps dirigés contre le panitumumab ont été évaluées par deux tests immunologiques différents (un test Elisa qui détecte les anticorps de forte affinité, et un test immunologique Biosensor qui détecte à la fois les anticorps de forte et de faible affinité). Les résultats de ces tests ont montré une incidence globale faible d’une réponse anticorps dirigés contre le panitumumab après administration. Des anticorps avant administration ont été détectés chez 5 des 636 patients (< 1 %) et chez 16 des 635 patients (2,5 %) respectivement par les tests immunologiques Elisa et Biosensor. Des anticorps neutralisants après administration ont été détectés chez 1 patient sur 447 (0,2 %) et chez 7 patients sur 447 (1,6 %) respectivement par les tests immunologiques Elisa et Biosensor. En comparaison avec les patients n’ayant pas développé d’anticorps, aucun lien n’a été observé entre la présence d’anticorps dirigés contre le panitumumab et la pharmacocinétique, l’efficacité et la tolérance du traitement.
- La détection de la formation d’anticorps dépend de la sensibilité et de la spécificité des tests. L’incidence observée d’un test positif aux anticorps peut être influencée par plusieurs facteurs dont la manipulation de l’échantillon, les traitements concomitants et la maladie sous-jacente. Par conséquent, la comparaison entre les incidences d’anticorps dirigés contre les autres produits peut induire en erreur.
- Efficacité clinique :
- L’efficacité de Vectibix chez les patients atteints de cancer colorectal métastatique (CCRm) avec progression de la maladie pendant ou après la chimiothérapie a été étudiée lors d’un essai clinique contrôlé randomisé (463 patients) et lors d’essais ouverts à un seul bras (384 patients). La tolérance chez les patients atteints de CCRm et recevant au moins une dose de Vectibix a été évaluée chez 920 patients. Des études complémentaires ont été réalisées avec Vectibix en monothérapie, chez des patients atteints d’autres tumeurs solides, en association avec une chimiothérapie avec ou sans bévacizumab chez les patients atteints de CCRm et en association avec une chimiothérapie chez les patients atteints de cancer du poumon non à petites cellules.
- Un essai international, randomisé, contrôlé, a été réalisé chez 463 patients atteints de cancer colorectal métastatique exprimant l’EGFR après échec confirmé des protocoles incluant de l’oxaliplatine et de l’irinotécan. Les patients ont été randomisés 1 : 1 afin de recevoir soit Vectibix à une dose de 6 mg/kg une fois toutes les deux semaines plus des soins palliatifs (SP), hors chimiothérapie, soit des soins palliatifs seuls. Les patients étaient traités jusqu’à progression de la maladie ou apparition d’une toxicité inacceptable. Dès progression de la maladie, les patients avec SP seuls étaient éligibles pour passer vers une étude complémentaire et recevoir Vectibix à une dose de 6 mg/kg une fois toutes les deux semaines.
- Sur les 463 patients, 63 % étaient de sexe masculin. L’âge médian était de 62 ans (de 27 à 83 ans), et 99 % étaient caucasiens. 396 patients (86 %) avaient un indice de performance ECOG de 0 ou 1. 67 % des patients étaient atteints de cancer du côlon et 33 % d’un cancer du rectum.
- Le critère principal a été la survie sans progression (SSP). Au cours d’une analyse d’ajustement des biais potentiels à partir d’évaluations non programmées, le taux de progression de la maladie ou de mortalité chez les patients traités par Vectibix a été réduit de 40 % par rapport aux patients traités par SP (risque relatif = 0,60 [IC 95 % : 0,49 ; 0,74], log-rank stratifié, p < 0,0001). Aucune différence de durée médiane de survie sans progression n’a été observée, plus de 50 % des patients ayant progressé avant la première visite programmée dans les deux groupes de traitement. Les taux de survie sans progression à la première visite programmée (semaine 8) étaient de 45,5 % pour le bras Vectibix plus SP et de 24,6 % pour le bras SP seuls, soit une différence de 20,9 % (IC 95 % : 12,4 ; 29,4). Aucune différence de la survie globale n’a été observée. Ceci peut être lié à l’administration de panitumumab après progression de la maladie des patients initialement randomisés dans le bras SP. La réponse tumorale déterminée selon les critères d’évaluation RECIST modifiés a été évaluée par un comité de revue centralisé. Dans l’ensemble, 9,5 % (IC 95 % : 6,1 ; 14,1) des patients recevant Vectibix plus SP, et 0 % (IC 95 % : 0,0 ; 1,6) des patients recevant SP seuls ont présenté une réponse objective confirmée (réponse partielle), avec une maladie stationnaire chez respectivement 26 % et 10 % des patients. Parmi les 176 patients traités par Vectibix suite à la progression de la maladie sous SP seuls, le taux de réponse (évaluation de l’investigateur) a été de 11,4 % (IC 95 % : 7,1 ; 17,0).
- La relation entre la mutation KRAS présentée dans le tissu tumoral conservé dans de la paraffine et le résultat clinique a été évaluée lors d’une analyse rétrospective.
- La présence des sept plus fréquentes mutations constitutives des codons 12 et 13 (Gly12Asp, Gly12Ala, Gly12Val, Gly12Ser, Gly12Arg, Gly12Cys et Gly13Asp) du gène KRAS a été analysée en utilisant une réaction en chaîne de la polymérase (PCR) allèle-spécifique. Cette analyse a été faite à partir d’échantillons de tumeur obtenus lors de la première résection de cancer colorectal. Parmi les 427 patients (92 %) évaluables pour le statut KRAS, 184 présentaient des mutations. Au cours d’une analyse d’ajustement des biais potentiels à partir d’évaluations non programmées, le risque relatif de la SSP a été de 0,49 (IC 95 % : 0,37 ; 0,65) en faveur du panitumumab dans le groupe KRAS non muté et de 1,070 (IC 95 % : 0,77 ; 1,48) dans le groupe KRAS muté. La différence de SSP médiane dans le groupe KRAS non muté a été de 8 semaines. Le taux de survie sans progression à la première visite programmée (semaine 8) dans le groupe KRAS non muté a été de 59,7 % pour le bras Vectibix plus SP et de 21,0 % pour le bras SP seuls, soit une différence de 38,7 % (IC 95 % : 27,4 ; 50,0). La différence de SSP médiane dans le groupe KRAS muté a été de 0 semaine. Le taux de survie sans progression à la première visite programmée (semaine 8) dans le groupe KRAS muté a été de 21,4 % dans le bras Vectibix plus SP et de 28,0 % dans le bras SP seuls, soit une différence de – 6,6 % (IC 95 % : – 19,0 ; 5,9). Aucune différence de la survie globale n’a été observée entre les deux groupes. Dans le groupe KRAS non muté, le taux de réponse a été de 17 % dans le bras panitumumab et de 0 % dans le bras SP. Dans le groupe KRAS muté, aucune réponse n’a été observée dans aucun des deux bras de traitement. Les taux de stabilisation de la maladie du groupe KRAS non muté ont été de 34 % dans le bras panitumumab et de 12 % dans le bras SP. Les taux de stabilisation de la maladie du groupe KRAS muté ont été de 12 % dans le bras panitumumab et de 8 % dans le bras SP. Le taux de réponse (selon l’évaluation de l’investigateur) des patients qui, après progression dans le bras SP seuls, étaient passés au panitumumab a été de 22 % (IC 95 % : 14,0 ; 31,9) dans le groupe KRAS non muté et de 0 % (IC 95 % : 0,0 ; 4,3) dans le groupe muté.
-
SSP* – Patient sans mutation KRAS (KRAS non muté) : 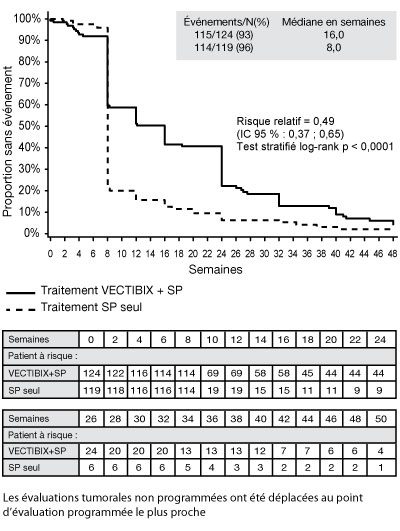
-
*
Survie sans progression
- Étude PACCE : dans cette étude randomisée, contrôlée, en ouvert, une chimiothérapie (oxaliplatine ou irinotécan) et du bévacizumab ont été administrés avec ou sans panitumumab, en traitement de première ligne des patients atteints de cancer colorectal métastatique (n = 1053 [n = 823 dans la cohorte oxaliplatine, n = 230 dans la cohorte irinotécan]). Le traitement par panitumumab a été interrompu en raison d’une réduction significative de la survie sans progression chez les patients du bras panitumumab, observée à l’occasion d’une analyse intermédiaire. L’objectif principal de l’étude était la comparaison de la SSP dans la cohorte oxaliplatine. Lors de l’analyse finale, le risque relatif de SSP était de 1,27 (IC 95 % : 1,06 ; 1,52). La médiane de SSP était de 10,0 mois (IC 95 % : 8,9 ; 11,0) et de 11,4 mois (IC 95 % : 10,5 ; 11,9) respectivement dans les bras avec et sans panitumumab. Une augmentation de la mortalité dans le bras panitumumab a été observée. Le risque relatif pour la survie globale était de 1,43 (IC 95 % : 1,11 ; 1,83). La médiane de survie globale était de 19,4 (IC 95 % : 18,4 ; 20,8) et de 24,5 (IC 95 % : 20,4 ; 24,5) respectivement dans les bras avec et sans panitumumab.
- Une analyse complémentaire des données d’efficacité en fonction du statut KRAS n’a pas révélé de sous-groupe de patients bénéficiant de l’association de Vectibix aux chimiothérapies à base d’oxaliplatine ou d’irinotécan et de bévacizumab. Pour les patients présentant un gène KRAS de type sauvage et faisant partie de la cohorte oxaliplatine, le risque relatif de SSP était de 1,36 (IC 95 % : 1,04 ; 1,77). Pour les patients présentant un gène KRAS muté, le risque relatif de SSP était de 1,25 avec (IC 95 % : 0,91 ; 1,71). Les résultats de la survie globale en faveur du bras de contrôle ont été observés chez les patients présentant un gène KRAS type sauvage appartenant à la cohorte oxaliplatine (risque relatif : 1,89 [IC 95 % : 1,30 ; 2,75]). Une tendance à la dégradation du taux de survie a également été observée avec le panitumumab dans la cohorte de l’irinotécan, indépendamment du statut mutationnel KRAS. Globalement, le traitement par panitumumab combiné aux chimiothérapies standard et le bévacizumab est associé à un profil bénéfice-risque défavorable, quel que soit le statut mutationnel du gène KRAS.
- Ce médicament a obtenu une autorisation de mise sur le marché conditionnelle. Cela signifie que des données complémentaires sur ce médicament sont attendues, en particulier des données confirmant l’effet chez les patients présentant une tumeur KRAS non mutée, qui repose actuellement sur une analyse rétrospective. Des données supplémentaires sont également attendues concernant les effets sur la SSP du panitumumab en association avec la chimiothérapie chez les patients présentant une tumeur KRAS non mutée. Des études évaluant cet effet sont actuellement en cours. Chaque année, l’agence européenne des médicaments (EMEA) évaluera les nouvelles données et le résumé des caractéristiques du produit (RCP) sera mis à jour si nécessaire.
PHARMACOCINÉTIQUE |
Vectibix administré en monothérapie ou en association avec une chimiothérapie présente des propriétés pharmacocinétiques non linéaires.
Après une administration unique de panitumumab en perfusion d’une heure, l’aire sous la courbe (ASC) a été augmentée plus que proportionnellement à la dose. La clairance (CL) du panitumumab est descendue de 30,6 à 4,6 ml/jour/kg pour des doses augmentées de 0,75 à 9 mg/kg. Cependant, à des doses supérieures à 2 mg/kg, l’ASC du panitumumab augmente presque proportionnellement à la dose.
Après administration de la dose recommandée (6 mg/kg une fois toutes les 2 semaines pendant une perfusion d’une heure), les concentrations de panitumumab ont atteint leur état d’équilibre après la troisième injection avec un pic moyen (± DS) et un point minimal respectivement de 213 µg/ml ± 59 et de 39 µg/ml ± 14. L’ASC moyenne (± DS) et la clairance ont été respectivement de 1306 µg/jour/ml ± 374 et de 4,9 ml/kg/jour ± 1,4. La demi-vie d’élimination a été approximativement de 7,5 jours (3,6 à 10,9 jours).
Une analyse pharmacocinétique de population a été effectuée afin d’étudier les effets potentiels de covariables sélectionnées sur les propriétés pharmacocinétiques du panitumumab. Les résultats suggèrent que l’âge (de 21 à 88 ans), le sexe, l’ethnie, les fonctions hépatiques et rénales, les agents chimiothérapeutiques et l’intensité de coloration de l’EGFR (1+, 2+, 3+) sur la membrane des cellules tumorales n’ont eu aucun impact observable sur la pharmacocinétique du panitumumab.
Chez les patients atteints d’insuffisance rénale ou hépatique, aucune étude pharmacocinétique du panitumumab n’a été réalisée.
SÉCURITE PRÉCLINIQUE |
Les effets secondaires observés chez l’animal à des niveaux d’exposition similaires à l’exposition clinique et présentant un intérêt clinique potentiel ont été : les rashs et les diarrhées, principales observations des études de toxicité à doses répétées, allant jusqu’à 26 semaines, sur des singes Cynomolgus. Ces résultats ont été observés à des doses approximativement équivalentes à celle recommandée chez l’homme et ont été réversibles à l’arrêt de l’administration du panitumumab. Les rashs et les diarrhées observés chez les singes sont considérés comme reliés à l’action pharmacologique du panitumumab et sont cohérents avec les toxicités observées avec d’autres inhibiteurs anti-EGFR.
Aucune étude de mutagenèse et carcinogenèse du panitumumab n’a été effectuée.
Les études animales ne sont pas suffisantes pour déterminer les effets délétères sur le développement embryofoetal, les niveaux d’exposition foetale au panitumumab n’ayant pas été étudiés. Le panitumumab a été responsable d’avortements et de morts foetales chez le singe Cynomolgus lors de son administration pendant la période d’organogenèse, à des doses approximativement équivalentes à la dose recommandée chez l’homme.
Aucune étude fondamentale sur la fertilité masculine n’a été menée. Néanmoins, l’observation microscopique des organes de reproduction masculins lors d’études de toxicité à doses répétées chez le singe Cynomolgus, à des doses approximativement 5 fois supérieures à la dose humaine exprimées en mg/kg, n’a révélé aucune différence avec les singes mâles contrôle. Les études de fertilité chez les femelles Cynomolgus ont montré que le panitumumab est susceptible d’allonger les cycles menstruels et/ou de provoquer des aménorrhées et de diminuer le taux de grossesses survenues pour toutes les doses évaluées.
Aucune étude animale du panitumumab sur le développement pré et postnatal n’a été menée. Avant de commencer le traitement par Vectibix, les patients devront être prévenus des risques potentiels du panitumumab sur le développement pré et postnatal.
INCOMPATIBILITÉS |
Ce médicament ne doit pas être mélangé avec d’autres médicaments, à l’exception de ceux cités en Modalités de manipulation/Élimination.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
A conserver au réfrigérateur (entre + 2 °C et + 8 °C). Ne pas congeler.
A conserver dans l’emballage extérieur d’origine à l’abri de la lumière.
- Après dilution :
- Vectibix ne contient aucun conservateur antimicrobien ou agent bactériostatique. Le produit doit être utilisé immédiatement après dilution. S’il n’est pas utilisé immédiatement, la durée et les conditions de stockage avant l’utilisation relèvent de la responsabilité de l’utilisateur et ne doivent pas dépasser 24 heures entre + 2 °C et + 8 °C. Ne pas congeler la solution diluée.
MODALITÉS MANIPULATION/ÉLIMINATION |
Vectibix doit être dilué dans une solution injectable de chlorure de sodium à 0,9 %, par un professionnel de santé dans des conditions d’asepsie. Ne pas secouer ni agiter vigoureusement le flacon. Ne pas administrer Vectibix si une décoloration est observée. Prélever la quantité nécessaire de Vectibix pour une dose de 6 mg/kg. Diluer dans un volume total de 100 ml. La concentration finale ne doit pas dépasser 10 mg/ml. Les doses supérieures à 1000 mg doivent être diluées dans 150 ml de solution injectable de chlorure de sodium à 0,9 % (cf Posologie et Mode d’administration). La solution diluée doit être mélangée délicatement par retournement, ne pas agiter.
Aucune incompatibilité n’a été observée entre Vectibix et la solution injectable de chlorure de sodium à 0,9 % dans les poches en PVC ou en polyoléfine.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament réservé à l’usage hospitalier. | |
| Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie. | |
| Médicament nécessitant une surveillance particulière pendant le traitement. | |
| AMM | EU/1/07/423/001 ; CIP 3400957181857 (RCP rév 13.12.2010) 5 ml. |
| EU/1/07/423/003 ; CIP 3400957182168 (RCP rév 13.12.2010) 20 ml. | |
| Collect. | |
| Prix ou tarif de responsabilité (HT) par UCD : | UCD 9307183 (flacon de 5 ml) : 430.00 euros. |
| UCD 9307177 (flacon de 20 ml) : 1720.00 euros. | |
| Inscrit sur la liste des spécialités prises en charge en sus des GHS. | |
AMGEN
62, bd Victor-Hugo. 92200 Neuilly-sur-Seine
Tél : 01 40 88 27 00. Fax : 01 40 88 27 90
Info médic et pharmacovigilance :
Tél : 09 69 36 33 63
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


