azacitidine
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p ml* | |
| Azacitidine (DCI) | 25 mg |
INDICATIONS |
- un syndrome myélodysplasique (SMD) de risque intermédiaire-2 ou élevé selon l’index pronostique international (International Prognostic Scoring System, IPSS),
- une leucémie myélomonocytaire chronique (LMMC) avec 10-29 % de blastes médullaires sans syndrome myéloprolifératif,
- une leucémie aiguë myéloblastique (LAM) avec 20-30 % de blastes et dysplasie de lignées multiples, selon la classification de l’Organisation mondiale de la santé (OMS).
POSOLOGIE ET MODE D’ADMINISTRATION |
La dose initiale recommandée pour le premier cycle de traitement, chez tous les patients, indépendamment des valeurs hématologiques de base, est de 75 mg/m2 de surface corporelle, par injection sous-cutanée, quotidiennement pendant 7 jours, suivis d’une période de repos de 21 jours (cycle de traitement de 28 jours).
Il est recommandé d’administrer au patient un minimum de 6 cycles de traitement. Le traitement doit être poursuivi tant qu’il apporte des bénéfices au patient ou jusqu’à progression de la maladie.
Le rapport réponse/toxicité hématologique et la toxicité rénale doivent être surveillés chez le patient (cf Mises en garde/Précautions d’emploi) ; il pourra être nécessaire de différer le début du cycle suivant ou de réduire la dose comme indiqué ci-dessous.
- Ajustement posologique lié à la toxicité hématologique :
- La toxicité hématologique se définit comme la numération sanguine la plus basse atteinte au cours d’un cycle donné (nadir) si les plaquettes chutent en dessous de 50,0 × 109/l et/ou que la numération des polynucléaires neutrophiles (PNN) chute en dessous de 1 × 109/l.
- La récupération correspond à une augmentation du nombre de cellules de la/des lignée(s) cellulaire(s) affectée(s) par la toxicité hématologique à hauteur d’au moins le nadir plus la moitié de la différence entre le nadir et la numération de base (soit : numération sanguine après récupération >= nadir + (0,5 × [numération de base – nadir]).
-
- Chez les patients dont les valeurs hématologiques de base ne sont pas diminuées (c’est-à-dire numération leucocytaire > 3,0 × 109/l et PNN > 1,5 × 109/l, et plaquettes > 75,0 × 109/l) avant initiation du traitement :
- Si une toxicité hématologique est observée suite au traitement par Vidaza, le cycle de traitement suivant par Vidaza doit être différé jusqu’à récupération de la numération plaquettaire et des PNN. Si la récupération est obtenue dans un délai de 14 jours, aucun ajustement posologique n’est nécessaire. En revanche, si la récupération ne se produit pas dans ce délai de 14 jours, la dose doit être réduite comme indiqué dans le tableau suivant. Après ces modifications posologiques, la durée du cycle sera ramenée à 28 jours.
-
Nadir des numérations % dose lors du cycle suivant, si la récupération* n’est pas obtenue dans les 14 jours PNN
(× 109/l)Plaquettes
(× 109/l)<= 1,0 <= 50,0 50 % > 1,0 > 50,0 100 % -
*
Récupération = numérations >= nadir + (0,5 × [numération de base – nadir]).
-
- Chez les patients dont les valeurs hématologiques de base sont diminuées (c’est-à-dire numération leucocytaire < 3,0 × 109/l ou PNN < 1,5 × 109/l ou plaquettes < 75,0 × 109/l) avant initiation du traitement :
- Si, suite au traitement par Vidaza, la réduction de la numération leucocytaire ou des PNN ou des plaquettes par rapport aux numérations antérieures au traitement est inférieure à 50 %, ou supérieure à 50 % mais qu’elle s’accompagne d’une amélioration d’une lignée cellulaire, le cycle suivant ne doit pas être différé et aucun ajustement posologique n’est requis.
- Si la réduction de la numération leucocytaire ou des PNN ou des plaquettes est supérieure à 50 % par rapport aux numérations antérieures au traitement mais ne s’accompagne de l’amélioration d’aucune lignée cellulaire, le cycle de traitement suivant par Vidaza doit être différé jusqu’à récupération de la numération plaquettaire et des PNN. Si la récupération est obtenue dans un délai de 14 jours, aucun ajustement posologique n’est nécessaire. En revanche, si la récupération ne se produit pas dans ce délai de 14 jours, la cellularité de la moelle osseuse doit être déterminée. Si la cellularité de la moelle osseuse est > 50 %, aucun ajustement posologique n’est requis. Si la cellularité de la moelle osseuse est <= 50 %, le traitement doit être différé et la dose réduite comme indiqué dans le tableau suivant :
-
Cellularité de la moelle osseuse % dose lors du cycle suivant, si la récupération* n’est pas obtenue dans les 14 jours Récupération* <= 21 jours Récupération* > 21 jours 15-50 % 100 % 50 % < 15 % 100 % 33 % -
*
Récupération = numérations >= nadir + (0,5 × [numération de base – nadir]).
- Après ces modifications posologiques, la durée du cycle sera ramenée à 28 jours.
- Populations particulières :
-
- Insuffisance rénale :
- Aucune étude formelle n’a été menée chez les patients atteints d’insuffisance rénale. En cas d’atteinte organique sévère, les patients doivent être surveillés attentivement afin de détecter les événements indésirables. Aucune modification spécifique de la dose initiale n’est recommandée chez les patients atteints d’insuffisance rénale (par exemple, niveau de base de créatinine sérique ou d’urée sanguine >= 2 fois la limite supérieure de la normale [LSN] ou bicarbonate sérique inférieur à 20 mmol/l) avant le début du traitement ; les ajustements posologiques ultérieurs devront se faire sur la base des valeurs hématologiques et des bilans rénaux. En cas de diminution inexpliquée du taux de bicarbonate sérique en dessous de 20 mmol/l, la dose doit être réduite de 50 % lors du cycle suivant. En cas d’augmentation inexpliquée de la créatinine sérique ou de l’urée sanguine à hauteur de >= 2 fois la valeur de base et la LSN, le cycle suivant doit être différé jusqu’à ce que les valeurs reviennent à la normale ou à leur niveau de base et la dose doit être réduite de 50 % lors du cycle de traitement suivant (cf Mises en garde/Précautions d’emploi).
-
- Insuffisance hépatique :
- Aucune étude formelle n’a été menée chez les patients atteints d’insuffisance hépatique (cf Mises en garde/Précautions d’emploi). En cas d’insuffisance hépatique sévère, les patients doivent être surveillés attentivement afin de détecter les événements indésirables. Aucune modification spécifique de la dose initiale n’est recommandée chez les patients atteints d’insuffisance hépatique avant le début du traitement ; les ajustements posologiques ultérieurs devront se faire sur la base des valeurs hématologiques. Vidaza est contre-indiqué chez les patients atteints de tumeurs hépatiques malignes à un stade avancé (cf Contre-indications, Mises en garde/Précautions d’emploi).
-
- Patients âgés :
- Aucun ajustement posologique spécifique n’est recommandé chez les patients âgés. La probabilité d’une insuffisance rénale étant plus importante chez les patients âgés, il pourra être utile de contrôler la fonction rénale.
-
- Enfants et adolescents :
- Ce médicament ne doit pas être utilisé chez l’enfant en dessous de 18 ans compte tenu de l’insuffisance de données concernant la sécurité et l’efficacité.
- Analyses de laboratoire :
- Un bilan hépatique et une mesure de la créatinine sérique doivent être effectués avant de commencer le traitement et avant chaque cycle de traitement. Une numération sanguine complète doit être réalisée avant de commencer le traitement et, si nécessaire, pour contrôler la réponse et la toxicité, mais dans tous les cas, au minimum avant chaque cycle de traitement.
Mode d’administration :
Une fois reconstitué, Vidaza doit être injecté par voie sous-cutanée dans le haut du bras, la cuisse ou l’abdomen. Les sites d’injection doivent être alternés. Chaque nouvelle injection doit être pratiquée à au moins 2,5 cm de distance du site précédent et en aucun cas sur une zone sensible, présentant une ecchymose, une rougeur ou une induration.
Des instructions détaillées sur la procédure de reconstitution et d’administration de Vidaza sont fournies dans la rubrique Modalités manipulation/élimination.
CONTRE-INDICATIONS |
- Hypersensibilité connue à la substance active ou à l’un des excipients.
- Tumeur hépatique maligne à un stade avancé (cf Mises en garde/Précautions d’emploi).
- Allaitement (cf Grossesse/Allaitement).
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- Toxicité hématologique :
- Le traitement par l’azacitidine est associé à des cas d’anémie, de neutropénie et de thrombocytopénie, en particulier au cours des 2 premiers cycles (cf Effets indésirables). Une numération sanguine complète doit être réalisée si nécessaire pour contrôler la réponse et la toxicité, mais dans tous les cas, au moins avant chaque cycle de traitement. Après administration de la dose recommandée pour le premier cycle, la dose utilisée lors des cycles suivants devra être réduite ou son administration différée en fonction du nadir des numérations et de la réponse hématologique (cf Posologie/Mode d’administration). Il devra être conseillé aux patients de signaler rapidement tout épisode fébrile. Il est également conseillé aux patients et à leurs médecins d’être attentifs aux signes et symptômes d’hémorragie.
- Insuffisance hépatique :
- Aucune étude formelle n’a été menée chez les patients atteints d’insuffisance hépatique. Chez les patients présentant une charge tumorale élevée due à une atteinte métastatique, de rares cas de coma hépatique progressif et de décès sous traitement par l’azacitidine ont été signalés, en particulier lorsque le taux de base d’albumine sérique de ces patients était < 30 g/l. L’azacitidine est contre-indiquée chez les patients atteints de tumeurs hépatiques malignes à un stade avancé (cf Contre-indications).
- Insuffisance rénale :
- Des anomalies rénales allant de l’augmentation du taux de créatinine sérique à l’insuffisance rénale et au décès ont été signalées dans de rares cas chez des patients traités par l’azacitidine en intraveineuse en association avec d’autres agents chimiothérapeutiques. Par ailleurs, une acidose tubulaire rénale définie par la chute du bicarbonate sérique à < 20 mmol/l associée à une urine alcaline et à une hypokaliémie (potassium sérique < 3 mmol/l), est survenue chez 5 sujets atteints de leucémie myéloïde chronique (LMC) traités par l’azacitidine et l’étoposide. En cas de diminution inexpliquée du bicarbonate sérique (< 20 mmol/l) ou d’augmentation de la créatinine sérique ou de l’urée sanguine, la dose doit être réduite ou son administration différée (cf Posologie/Mode d’administration).
- En cas d’insuffisance rénale, les patients doivent être surveillés attentivement afin de détecter toute toxicité car l’azacitidine et/ou ses métabolites sont excrétés principalement par les reins (cf Posologie/Mode d’administration).
- Affections cardiaque et pulmonaire :
- La sécurité d’emploi et l’efficacité de Vidaza n’ont pas été établies chez les patients présentant des antécédents d’insuffisance cardiaque congestive sévère, d’affection cardiaque cliniquement instable ou d’affection pulmonaire, ces patients ayant été exclus de l’étude clinique pivot.
INTERACTIONS |
GROSSESSE et ALLAITEMENT |
Il n’existe pas de données suffisamment pertinentes concernant l’utilisation de l’azacitidine chez la femme enceinte. Des études effectuées chez la souris ont mis en évidence une toxicité sur la reproduction (cf Sécurité préclinique). Le risque potentiel en clinique n’est pas connu. Étant donnés les résultats des études chez l’animal et son mécanisme d’action, l’azacitidine ne doit pas être utilisée pendant la grossesse, en particulier pendant le premier trimestre, à moins d’une nécessité clairement établie. Les effets bénéfiques du traitement doivent être évalués au cas par cas au regard des risques éventuels encourus par le foetus.
Les femmes en âge de procréer et les hommes doivent utiliser une contraception efficace pendant le traitement et jusqu’à 3 mois après l’arrêt du traitement.
Fertilité :
Il n’existe pas de données concernant les effets de l’azacitidine sur la fertilité humaine. Chez l’animal, des effets indésirables de l’azacitidine sur la fertilité des mâles ont été décrits (cf Sécurité préclinique). Il devra être déconseillé aux hommes de concevoir pendant le traitement et ils devront utiliser une contraception efficace pendant le traitement et jusqu’à 3 mois après l’arrêt du traitement. Avant de commencer le traitement, il est conseillé aux patients de sexe masculin de se renseigner sur les procédures de conservation du sperme.
Allaitement :
On ignore si l’azacitidine ou ses métabolites sont excrétés dans le lait maternel humain. Étant données les réactions indésirables graves possibles chez l’enfant allaité, l’allaitement est contre-indiqué pendant le traitement par l’azacitidine.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
| Classe de systèmes d’organes | Effets indésirables |
| Infections et infestations | Très fréquent : pneumonie, rhinopharyngite. |
| Fréquent : septicémie sur neutropénie, infection des voies respiratoires supérieures, infection urinaire, sinusite, pharyngite, rhinite, herpès. | |
| Affections hématologiques et du système lymphatique | Très fréquent : neutropénie fébrile, neutropénie, leucopénie, thrombocytopénie, anémie. |
| Fréquent : aplasie médullaire, pancytopénie. | |
| Affections du système immunitaire | Peu fréquent : réactions d’hypersensibilité. |
| Troubles du métabolisme et de la nutrition | Très fréquent : anorexie. |
| Fréquent : hypokaliémie. | |
| Affections psychiatriques | Fréquent : état confusionnel, anxiété, insomnies. |
| Affections du système nerveux | Très fréquent : vertiges, céphalées. |
| Fréquent : hémorragie intracrânienne, léthargie. | |
| Affections oculaires | Fréquent : hémorragie oculaire, hémorragie conjonctivale. |
| Affections vasculaires | Fréquent : hypertension, hypotension, hématome. |
| Affections respiratoires, thoraciques et médiastinales | Très fréquent : dyspnée. |
| Fréquent : dyspnée d’effort, douleur pharyngolaryngée. | |
| Affections gastro-intestinales | Très fréquent : diarrhée, vomissements, constipation, nausées, douleurs abdominales. |
| Fréquent : hémorragie gastro-intestinale, hémorragie hémorroïdaire, stomatite, hémorragie gingivale, dyspepsie. | |
| Affections de la peau et du tissu sous-cutané | Très fréquent : pétéchies, prurit, éruption cutanée, ecchymoses. |
| Fréquent : purpura, alopécie, érythème, éruption maculaire. | |
| Affections musculo-squelettiques et systémiques | Très fréquent : arthralgie. |
| Fréquent : myalgie, douleurs musculo-squelettiques. | |
| Affections du rein et des voies urinaires | Fréquent : hématurie. |
| Troubles généraux et anomalies au site d’administration | Très fréquent : fatigue, pyrexie, douleurs thoraciques, érythème au site d’injection, douleur au site d’injection, réaction (non précisée) au site d’injection. |
| Fréquent : site d’injection : ecchymose, hématome, induration, éruption cutanée, prurit, inflammation, décoloration, nodule et hémorragie ; malaise. | |
| Investigations | Fréquent : perte de poids. |
- Réactions hématologiques indésirables :
- Les réactions indésirables signalées le plus fréquemment en association avec le traitement par l’azacitidine ont été les réactions hématologiques, notamment les thrombocytopénies, les neutropénies et les leucopénies, généralement de grade 3 ou 4. Le risque de survenue de ces événements est plus important pendant les 2 premiers cycles, après quoi ils deviennent moins fréquents chez le patient dont la fonction hématologique se rétablit. Dans la plupart des cas, les réactions hématologiques indésirables ont été prises en charge par le biais d’une surveillance régulière des numérations sanguines complètes et, si nécessaire, en différant l’administration de l’azacitidine lors du cycle suivant, à l’aide d’une prophylaxie antibiotique et/ou d’un traitement de support par facteur de croissance (G-CSF, par exemple) pour la neutropénie et de transfusions pour l’anémie ou la thrombocytopénie.
- Infections :
- L’insuffisance médullaire peut aboutir à une neutropénie et un risque accru d’infection. Des réactions indésirables graves, telles que des septicémies sur neutropénie (0,8 %) et des pneumonies (2,5 %), ont été signalées chez des patients recevant de l’azacitidine. Les infections peuvent être prises en charge en utilisant des agents anti-infectieux associés à un traitement de support par facteur de croissance (G-CSF, par exemple) pour la neutropénie.
- Hémorragies :
- Des hémorragies peuvent se produire chez les patients sous azacitidine. Des réactions indésirables graves telles que des hémorragies gastro-intestinales (0,8 %) et des hémorragies intracrâniennes (0,5 %) ont été rapportées. Les patients doivent être surveillés afin de détecter les signes et symptômes d’hémorragie, en particulier en cas de thrombocytopénie préexistante ou liée au traitement.
- Hypersensibilité :
- De graves réactions d’hypersensibilité (0,25 %) ont été décrites chez des patients sous azacitidine. En cas de réaction de type anaphylactique, le traitement par l’azacitidine doit être immédiatement interrompu et un traitement symptomatique adapté doit être instauré.
- Réactions indésirables cutanées et du tissu sous-cutané :
- En majorité, les réactions cutanées et sous-cutanées indésirables concernent le site d’injection. Aucune de ces réactions indésirables n’a nécessité d’interrompre provisoirement ou définitivement le traitement par l’azacitidine ou de réduire la dose d’azacitidine lors de l’étude pivot. Les réactions indésirables sont survenues majoritairement au cours des 2 premiers cycles et ont eu tendance à diminuer lors des cycles suivants. Les réactions sous-cutanées indésirables telles que éruption/inflammation/prurit au site d’injection, éruption cutanée, érythème et lésion cutanée peuvent nécessiter un traitement concomitant par des antihistaminiques, des corticostéroïdes et des anti-inflammatoires non stéroïdiens (AINS) notamment.
- Réactions indésirables gastro-intestinales :
- Les réactions gastro-intestinales indésirables signalées le plus fréquemment en association avec le traitement par l’azacitidine ont été notamment la constipation, la diarrhée, les nausées et les vomissements. Ces réactions indésirables ont été prises en charge à l’aide d’un traitement symptomatique par des anti-émétiques pour les nausées et les vomissements, des anti-diarrhéiques pour la diarrhée et des laxatifs et/ou émollients fécaux pour la constipation.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : agents antinéoplasiques, analogues de la pyrimidine ; code ATC : L01BC07.
- Mécanisme d’action :
- L’azacitidine pourrait exercer ses effets antinéoplasiques par des mécanismes multiples comprenant une cytotoxicité directe à l’encontre des cellules hématopoïétiques anormales de la moelle osseuse et une hypométhylation de l’ADN. Les effets cytotoxiques de l’azacitidine pourraient résulter de mécanismes multiples, comprenant l’inhibition de la synthèse de l’ADN, de l’ARN et de protéines, son incorporation dans l’ARN et l’ADN, et l’activation des voies de dégradation de l’ADN. Les cellules non prolifératives sont relativement insensibles à l’azacitidine. L’incorporation de l’azacitidine dans l’ADN entraîne l’inactivation des ADN méthyltransférases, ce qui engendre une hypométhylation de l’ADN. L’hypométhylation de l’ADN des gènes présentant une méthylation aberrante impliqués dans la régulation, la différenciation et les voies de destruction du cycle cellulaire normal peut entraîner une ré-expression des gènes et une restauration des fonctions suppressives des cellules cancéreuses. L’importance relative de l’hypométhylation de l’ADN par rapport à la cytotoxicité ou aux autres activités de l’azacitidine en termes de résultats cliniques observés n’a pas été établie.
- Efficacité et sécurité clinique :
- L’efficacité et la sécurité d’emploi de Vidaza ont été étudiées par une étude comparative, internationale, multicentrique, contrôlée, ouverte, randomisée, sur groupes parallèles, de phase 3 (AZA PH GL 2003 CL 001) chez des patients atteints de : SMD à risque intermédiaire-2 ou élevé selon l’index pronostique international (International Prognostic Scoring System, IPSS), anémie réfractaire avec excès de blastes (AREB), anémie réfractaire avec excès de blastes en transformation (AREB-T) et leucémie myélomonocytaire chronique (LMMC) modifiée selon le système de classification franco-américano-britannique (FAB). L’AREB-T (21-30 % de blastes) est désormais classée parmi les LAM d’après le système de classification actuel de l’OMS. L’azacitidine ajoutée au traitement symptomatique optimal (Best Supportive Care, BSC) (n = 179) a été comparé aux traitements classiques (Conventional Care Regimens, CCR). Les CCR étaient constitués du BSC seul (n = 105), de cytarabine à faible dose plus le BSC (n = 49) ou d’une chimiothérapie d’induction standard plus le BSC (n = 25). Le choix d’1 des 3 CCR a été fait par le médecin pour chaque patient avant la randomisation. Les patients ont reçu le CCR préalablement sélectionné s’ils n’étaient pas randomisés dans le groupe de traitement par Vidaza. Entre autres critères d’inclusion, les patients devaient présenter un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0-2. Les patients atteints de SMD secondaire ont été exclus de l’étude. Le critère d’évaluation principal de l’étude était la durée de survie globale. Vidaza a été administré par injection sous-cutanée à la dose de 75 mg/m2 quotidiennement pendant 7 jours, suivis d’une période de repos de 21 jours (cycle de traitement de 28 jours) pendant une durée médiane de 9 cycles (intervalle : 1-39) et une durée moyenne de 10,2 cycles. Au sein de la population en intention de traiter (IdT), l’âge médian était de 69 ans (intervalle : 38 à 88 ans).
- Dans la population en IdT, constituée de 358 patients (179 sous azacitidine et 179 sous CCR), le traitement par Vidaza a été associé à une durée de survie médiane de 24,46 mois contre 15,02 mois chez les patients sous CCR, soit une différence de 9,4 mois, avec une valeur de p = 0,0001 avec le test de log-Rank stratifié. Le rapport de risque correspondant aux effets de ce traitement a été de 0,58 (IC à 95 % : 0,43 ; 0,77). Le taux de survie à deux ans a été de 50,8 % chez les patients sous azacitidine contre 26,2 % chez les patients sous CCR (p < 0,0001).
-
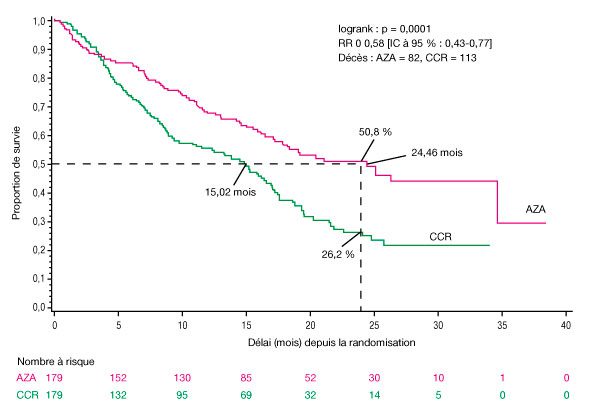
Abréviations : AZA = azacitidine ; CCR = traitements classiques (conventional care regimens) ; IC = intervalle de confiance ; RR = risque relatif.
- Les bénéfices de Vidaza en terme de survie ont été cohérents indépendamment du traitement CCR choisi (BSC seul, cytarabine à faible dose plus BSC ou chimiothérapie d’induction standard plus BSC) dans le groupe témoin.
- Lorsque les différents sous-groupes cytogénétiques IPSS ont été analysés, des résultats similaires ont été obtenus en termes de survie globale médiane dans tous les groupes (profils cytogénétiques de bon pronostic, de pronostic intermédiaire ou de mauvais pronostic y compris les monosomies 7).
- Les analyses des sous-groupes par classes d’âge ont fait apparaître une augmentation de la survie globale médiane dans tous les groupes (< 65 ans, >= 65 ans et >= 75 ans).
- Le traitement par Vidaza a été associé à un délai médian avant décès ou transformation en LAM de 13,0 mois contre 7,6 mois chez les patients sous CCR, soit une amélioration de 5,4 mois avec une valeur de p = 0,0025 avec le test de log-Rank stratifié.
- Le traitement par Vidaza a également été associé à une réduction des cytopénies et des symptômes associés. Le traitement par Vidaza a entraîné une réduction du besoin en transfusions de plaquettes et de globules rouges. Sur l’ensemble des patients du groupe sous azacitidine qui étaient initialement dépendants des transfusions de globules rouges, 45,0 % sont devenus indépendants des transfusions de globules rouges pendant la période de traitement, contre 11,4 % dans les groupes sous CCR poolés (soit une différence statistiquement significative [p < 0,0001] de 33,6 % [IC à 95 % : 22,4 ; 44,6]). Parmi les patients initialement dépendants des transfusions de globules rouges devenus indépendants, la durée médiane de cette indépendance a été de 13 mois dans le groupe sous azacitidine.
- La réponse au traitement a été évaluée par l’investigateur ou par le Comité de revue indépendant (CRI). La réponse globale (rémission complète [RC] + rémission partielle [RP]) déterminée par l’investigateur a été de 29 % dans le groupe sous azacitidine et de 12 % dans les groupes sous CCR poolés (p = 0,0001). La réponse globale (RC + RP) déterminée par le CRI dans l’étude AZA PH GL 2003 CL 001 a été de 7 % (12/179) dans le groupe sous azacitidine contre 1 % (2/179) dans les groupes sous CCR poolés (p = 0,0113). Les différences entre les évaluations du CRI et de l’investigateur s’expliquent par l’utilisation des critères du groupe de travail international (International Working Group, IWG), lesquels requièrent une amélioration des numérations sanguines périphériques et le maintien de cette amélioration pendant au minimum 56 jours. Un bénéfice en terme de survie a également été démontré chez les patients n’ayant pas obtenu de réponse complète/partielle suite au traitement par l’azacitidine. Une amélioration hématologique (majeure ou mineure) déterminée par le CRI a été obtenue chez 49 % des patients sous azacitidine contre 29 % des patients des groupes sous CCR poolés (p < 0,0001).
- Sur l’ensemble des patients présentant initialement une ou plusieurs anomalies cytogénétiques, le pourcentage de patients ayant bénéficié d’une réponse cytogénétique majeure a été similaire dans le groupe sous azacitidine et dans les groupes sous CCR poolés. Le taux de réponses cytogénétiques mineures a été supérieur de façon statistiquement significative (p = 0,0015) dans le groupe sous azacitidine (34 %) par rapport aux groupes sous CCR poolés (10 %).
PHARMACOCINÉTIQUE |
Les propriétés pharmacocinétiques de l’azacitidine ont été étudiées suite à l’administration de doses uniques de 75 mg/m2 par voie sous-cutanée et intraveineuse.
- Absorption :
- L’azacitidine a été rapidement absorbée suite à l’administration sous-cutanée, avec un pic de concentration plasmatique de 750 ± 403 ng/ml atteint 0,5 h (premier prélèvement effectué) après l’administration. La biodisponibilité absolue de l’azacitidine administrée par voie sous-cutanée par rapport à l’azacitidine administrée par voie intraveineuse a été d’approximativement 89 % d’après l’aire sous la courbe (ASC).
- Distribution :
- Suite à l’administration intraveineuse, le volume de distribution moyen a été de 76 ± 26 l et la clairance systémique de 147 ± 47 l/h.
- Métabolisme :
- D’après les données in vitro, le métabolisme de l’azacitidine ne semble pas être médié par les isoenzymes du cytochrome P450 (CYP), les UDP-glucuronosyl-transférases (UGT), les sulfotransférases (SULT) et les glutathion transférases (GST).
- L’azacitidine est métabolisée par hydrolyse spontanée et par désamination par la cytidine désaminase. Au niveau des fractions hépatiques humaines S9, la formation des métabolites a été indépendante du NADPH, ce qui implique que le métabolisme est dans tous les cas catalysé par les enzymes cytosoliques. Les études in vitro de l’azacitidine sur des cultures d’hépatocytes humains indiquent que, à des concentrations de 1,0 µM à 100 µM (c’est-à-dire jusqu’à 30 fois environ la concentration atteinte en pratique clinique), l’azacitidine n’a pas d’effet inducteur sur les isoenzymes du cytochrome P450 (CYP) 1A2, 2C19, 3A4 ou 3A5. Lors d’une étude visant à évaluer l’inhibition d’une série d’isoenzymes du P450 (CYP 1A2, 2C9, 2C19, 2D6, 2E1 et 3A4) incubées avec 100 µM d’azacitidine, les valeurs de la CI50 n’ont pas pu être déterminées ; il est donc improbable que l’azacitidine entraîne une inhibition enzymatique aux concentrations plasmatiques atteintes en pratique clinique. La capacité du produit à inhiber le CYP2B6 ou 2C8 n’a pas été étudiée.
- Excrétion :
- L’azacitidine est rapidement éliminée du plasma, avec une demi-vie d’élimination (t½) moyenne, après administration sous-cutanée, de 41 ± 8 minutes. L’élimination de l’azacitidine et/ou de ses métabolites se fait principalement par excrétion urinaire. Après administration intraveineuse et sous-cutanée de 14C-azacitidine, 50-85 % de la radioactivité administrée a été retrouvée dans les urines contre < 1 % dans les selles.
- Populations particulières :
- Les effets de l’insuffisance rénale ou hépatique (cf Posologie/Mode d’administration), du sexe, de l’âge ou des origines ethniques sur les propriétés pharmacocinétiques de l’azacitidine n’ont pas été formellement étudiés.
- Caractéristiques pharmacogénomiques :
- Les effets des polymorphismes connus de la cytidine désaminase sur le métabolisme de l’azacitidine n’ont pas été formellement étudiés.
SÉCURITE PRÉCLINIQUE |
L’azacitidine induit des mutations génétiques et des aberrations chromosomiques dans les systèmes cellulaires de bactéries et de mammifères in vitro. La carcinogénicité de l’azacitidine a été évaluée chez la souris et le rat. L’azacitidine a induit des tumeurs dans le système hématopoïétique des souris femelles, lorsqu’elle a été administrée par voie intrapéritonéale 3 fois par semaine pendant 52 semaines. Une augmentation de l’incidence des tumeurs du système lymphoréticulaire, des poumons, des glandes mammaires et de la peau a été observée chez les souris traitées par l’azacitidine administrée par voie intrapéritonéale pendant 50 semaines. Une étude du potentiel tumorigène chez le rat a fait apparaître une augmentation de l’incidence des tumeurs testiculaires.
Les études d’embryotoxicité précoce chez la souris ont révélé une fréquence de 44 % des décès embryonnaires intra-utérins (résorption accrue) suite à une injection unique d’azacitidine par voie intrapéritonéale en cours d’organogenèse. Des anomalies du développement cérébral ont été détectées chez la souris sous azacitidine pendant ou avant la soudure de la voûte palatine. Chez le rat, l’azacitidine n’a provoqué aucun effet indésirable lorsqu’elle était administrée avant l’implantation mais elle s’est révélée clairement embryotoxique lorsqu’elle était administrée pendant l’organogenèse. Les anomalies foetales survenues en cours d’organogenèse ont été notamment : anomalies du SNC (exencéphalie/encéphalocèle), anomalies des membres (micromélie, pied bot, syndactylie, oligodactylie) et autres (micrognathie, laparoschisis, oedème et anomalies costales).
L’administration d’azacitidine chez la souris mâle avant l’accouplement avec une femelle non traitée a entraîné une réduction de la fertilité et des pertes dans la descendance lors du développement embryonnaire et post-natal ultérieur. Le traitement des rats mâles a engendré une réduction de la masse testiculaire et des épididymites, une diminution de la numération des spermatozoïdes, une réduction du taux de grossesses, une augmentation du nombre d’embryons anormaux et des pertes d’embryons accrues chez les femelles fécondées (cf Mises en garde/Précautions d’emploi).
INCOMPATIBILITÉS |
Ce médicament ne doit pas être mélangé avec d’autres médicaments à l’exception de ceux mentionnés dans la rubrique Modalités manipulation/élimination.
CONDITIONS DE CONSERVATION |
Après reconstitution : la durée de stabilité chimique et physique démontrée à l’utilisation pour le médicament reconstitué est de 45 minutes à 25 °C et de 8 heures entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement. Dans le cas contraire, à l’utilisation, la durée et les conditions de conservation avant administration sont sous la responsabilité de l’utilisateur mais ne doivent en aucun cas dépasser 8 heures entre 2 °C et 8 °C.
MODALITÉS MANIPULATION/ÉLIMINATION |
- Recommandations pour une manipulation en toute sécurité :
- Vidaza est un médicament cytotoxique et, comme pour toute autre substance potentiellement toxique, la manipulation et la préparation de la suspension d’azacitidine doivent être réalisées avec précaution. Les procédures appropriées de manipulation et d’élimination applicables aux médicaments anticancéreux doivent être respectées.
- Si l’azacitidine reconstituée entre en contact avec la peau, rincer immédiatement et abondamment avec de l’eau et du savon. Si elle entre en contact avec les muqueuses, rincer abondamment avec de l’eau.
- Procédure de reconstitution :
-
- Réunir les éléments suivants :
- flacon(s) d’azacitidine ; flacon(s) d’eau pour préparations injectables ; gants chirurgicaux non stériles ;
- lingettes désinfectantes ; seringue(s) pour injection de 5 ml avec aiguille(s).
- flacon(s) d’azacitidine ; flacon(s) d’eau pour préparations injectables ; gants chirurgicaux non stériles ;
- Aspirer 4 ml d’eau pour préparations injectables dans la seringue, en veillant à expulser toute bulle d’air présente dans la seringue.
- Introduire l’aiguille de la seringue contenant les 4 ml d’eau pour préparations injectables dans le bouchon en élastomère du flacon d’azacitidine et injecter l’eau pour préparations injectables dans le flacon.
- Retirer la seringue et l’aiguille, agiter vigoureusement le flacon jusqu’à obtenir une suspension trouble uniforme. Après reconstitution, chaque ml de suspension contient 25 mg d’azacitidine (100 mg/4 ml). Le produit reconstitué se présente sous la forme d’une suspension trouble homogène dépourvue d’agglomérats. Jeter la suspension si elle contient de grosses particules ou des agglomérats.
- Nettoyer le dessus du bouchon en élastomère et introduire une nouvelle seringue avec aiguille. Retourner le flacon et s’assurer que l’extrémité de l’aiguille se situe en dessous de la surface du liquide. Tirer le piston afin d’aspirer le volume de médicament correspondant à la dose appropriée, en veillant à expulser toute bulle d’air présente dans la seringue. Retirer la seringue avec aiguille du flacon et jeter l’aiguille.
- Fixer solidement une aiguille pour injection sous-cutanée neuve (calibre 25 recommandé) sur la seringue. Afin de réduire l’incidence des réactions locales au site d’injection, l’aiguille ne doit pas être purgée avant l’injection.
- Si nécessaire (doses supérieures à 100 mg), réitérer les étapes ci-dessus pour achever la préparation de la suspension. Si la dose est supérieure à 100 mg (4 ml), elle doit être répartie de façon égale dans 2 seringues (par exemple, pour une dose de 150 mg = 6 ml, 2 seringues de 3 ml chacune).
- Le contenu de la seringue doit être remis en suspension immédiatement avant l’administration. Au moment de l’injection, la température de la suspension doit atteindre environ 20 °C-25 °C. Pour remettre le produit en suspension, faire rouler la seringue vigoureusement entre les paumes de la main jusqu’à obtenir une suspension trouble uniforme. Jeter la suspension si elle contient de grosses particules ou des agglomérats.
- Réunir les éléments suivants :
- La suspension de Vidaza doit être préparée immédiatement avant utilisation et la suspension reconstituée doit être administrée dans les 45 minutes. Si ce délai de 45 minutes est dépassé, la suspension reconstituée doit être éliminée de façon appropriée et une nouvelle dose doit être préparée. Si toutefois le produit doit être reconstitué à l’avance, la dose doit être placée au réfrigérateur (entre 2 °C et 8 °C) immédiatement après reconstitution et y être conservée pendant 8 heures maximum. Si ce délai de 8 heures dans le réfrigérateur est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée. La seringue contenant la suspension reconstituée doit être laissée à température ambiante pendant 30 minutes avant l’administration jusqu’à ce qu’elle atteigne une température d’environ 20 °C-25 °C. Si ce délai de 30 minutes est dépassé, la suspension doit être éliminée de façon appropriée et une nouvelle dose doit être préparée.
- Calcul d’une dose spécifique :
- La dose totale basée sur la surface corporelle peut être calculée ainsi :
- Dose totale (mg) = dose (mg/m2) × surface corporelle (m2)
- Le tableau suivant est proposé uniquement à titre d’exemple pour montrer comment calculer une dose d’azacitidine spécifique pour une surface corporelle moyenne de 1,8 m2.
-
Dose en mg/m2
(% de la dose initiale recommandée)Dose totale basée sur une surface corporelle de 1,8 m2 Nombre de flacons nécessaires Volume total de suspension reconstituée requis 75 mg/m2 (100 %) 135 mg 2 flacons 5,4 ml 37,5 mg/m2 (50 %) 67,5 mg 1 flacon 2,7 ml 25 mg/m2 (33 %) 45 mg 1 flacon 1,8 ml
- Mode d’administration :
- Une fois reconstitué, Vidaza doit être injecté par voie sous-cutanée (introduire l’aiguille avec un angle de 45-90°) à l’aide d’une aiguille de calibre 25 dans le haut du bras, la cuisse ou l’abdomen.
- Les doses supérieures à 4 ml doivent être injectées dans deux sites différents.
- Les sites d’injection doivent être alternés. Chaque nouvelle injection doit être pratiquée à au moins 2,5 cm de distance du site précédent et en aucun cas sur une zone sensible, présentant une ecchymose, une rougeur ou une induration.
- Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament soumis à prescription hospitalière. | |
| Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie. | |
| Médicament nécessitant une surveillance particulière pendant le traitement. | |
| AMM | EU/1/08/488/001 ; CIP 3400939126586 (rév 20.10.2009). |
| Inscrit sur la liste de rétrocession, avec prise en charge à 100 %. Inscrit sur la liste des spécialités prises en charge en sus des GHS. Collect. |
Titulaire de l’AMM : Celgene, Europe Limited, Riverside House, Riverside Walk, Windsor, Berkshire, SL4 1NA, Royaume-Uni.
CELGENE
16-18, rue du Quatre-Septembre. 75002 Paris
Tél : 01 53 42 43 00. Fax : 01 53 42 43 20
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


