aprépitant
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p gélule | |
| Aprépitant (DCI) | 80 mg |
| ou | 125 mg |
Teneur en sucrose : 80 mg/gél 80 mg ; 125 mg/gél 125 mg.
INDICATIONS |
- Prévention des nausées et des vomissements aigus et retardés associés à une chimiothérapie anticancéreuse hautement émétisante comprenant du cisplatine chez l’adulte.
- Prévention des nausées et des vomissements associés à une chimiothérapie anticancéreuse moyennement émétisante chez l’adulte.
POSOLOGIE ET MODE D’ADMINISTRATION |
Emend est administré durant 3 jours dans le cadre d’un schéma thérapeutique comportant un corticostéroïde et un antagoniste 5-HT3. La posologie recommandée d’Emend est de 125 mg par voie orale (PO), une fois par jour, une heure avant le début de la chimiothérapie à J1 et de 80 mg PO une fois par jour à J2 et J3. Le fosaprépitant 115 mg, une prodrogue lyophilisée de l’aprépitant, peut être substitué à Emend 125 mg par voie orale, 30 minutes avant la chimiothérapie, uniquement le jour 1 du schéma thérapeutique pour la prévention des nausées et vomissement induits par la chimiothérapie (NVIC), et administré en perfusion pendant 15 minutes. Se référer au Résumé des caractéristiques du produit du fosaprépitant.
Coût du traitement journalier : 26,30 euro(s) (2 gél à 80 mg) ; 25,43 euro(s) (1 gél à 125 mg et 2 gél à 80 mg).Au cours des essais cliniques avec Emend, les schémas thérapeutiques suivants ont été utilisés pour la prévention des nausées et des vomissements associés à une chimiothérapie anticancéreuse émétisante.
| J1 | J2 | J3 | J4 | |
| Emend | 125 mg (voie orale) | 80 mg (voie orale) | 80 mg (voie orale) | – |
| Dexaméthasone | 12 mg (voie orale) | 8 mg (voie orale) | 8 mg (voie orale) | 8 mg (voie orale) |
| Ondansétron | 32 mg IV | – | – | – |
Emend a été administré par voie orale 1 heure avant le début de la chimiothérapie à J1 et le matin de J2 et de J3.
La dexaméthasone a été administrée 30 minutes avant le début de la chimiothérapie à J1 et le matin de J2 à J4. La dose de dexaméthasone a été déterminée en tenant compte des interactions médicamenteuses.
L’ondansétron a été administré par voie intraveineuse 30 minutes avant le début de la chimiothérapie à J1.
| J1 | J2 | J3 | |
| Emend | 125 mg (voie orale) | 80 mg (voie orale) | 80 mg (voie orale) |
| Dexaméthasone | 12 mg (voie orale) | – | – |
| Ondansétron par voie orale | 2 × 8 mg (voie orale) | – | – |
Emend a été administré par voie orale 1 heure avant le début de la chimiothérapie à J1 et le matin de J2 et de J3.
La dexaméthasone a été administrée 30 minutes avant le début de la chimiothérapie à J1. La dose de dexaméthasone a été déterminée en tenant compte des interactions médicamenteuses.
Une gélule d’ondansétron à 8 mg a été administrée 30 à 60 minutes avant le début de la chimiothérapie et une gélule à 8 mg a été administrée 8 heures après la première dose à J1.
Les données d’efficacité en association avec d’autres corticostéroïdes et d’autres antagonistes 5-HT3 sont limitées. Pour plus d’informations concernant la coadministration avec des corticostéroïdes, cf Interactions.
Se référer aux Résumés des caractéristiques du produit des agents antiémétiques coadministrés.
- Sujet âgé (>= 65 ans) :
- Aucun ajustement posologique n’est nécessaire chez le sujet âgé (cf Pharmacocinétique).
- Sexe :
- Aucun ajustement posologique n’est nécessaire en fonction du sexe (cf Pharmacocinétique).
- Insuffisance rénale :
- Aucun ajustement posologique n’est nécessaire chez les patients insuffisants rénaux, y compris chez les patients insuffisants rénaux au stade terminal et hémodialysés (cf Pharmacocinétique).
- Insuffisance hépatique :
- Aucun ajustement posologique n’est nécessaire chez les patients ayant une insuffisance hépatique légère. Les données disponibles chez les patients ayant une insuffisance hépatique modérée sont limitées, et aucune donnée chez les patients ayant une insuffisance hépatique sévère n’est disponible (cf Mises en garde et Précautions d’emploi, Pharmacocinétique).
- Enfant et adolescent :
- Il n’est pas recommandé d’utiliser Emend chez l’enfant de moins de 18 ans car les données concernant sa sécurité d’emploi et son efficacité sont insuffisantes.
Mode d’administration :
La gélule doit être avalée entière.
Emend peut être pris avec ou sans aliments.
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active ou à l’un des excipients.
- Coadministration avec le pimozide, la terfénadine, l’astémizole ou le cisapride (cf Interactions).
MISES EN GARDE et PRÉCAUTIONS D’EMPLOI |
- Les données disponibles chez les patients ayant une insuffisance hépatique modérée sont limitées, et aucune donnée chez les patients ayant une insuffisance hépatique sévère n’est disponible. Emend doit être utilisé avec précaution chez ces patients(cf Pharmacocinétique).
- Emend doit être utilisé avec précaution chez les patients prenant de façon concomitante par voie orale des substances actives métabolisées principalement par le CYP3A4 et ayant une marge thérapeutique étroite, telles que la ciclosporine, le tacrolimus, le sirolimus, l’évérolimus, l’alfentanil, la diergotamine, l’ergotamine, le fentanyl et la quinidine (cf Interactions). De plus, l’administration concomitante d’irinotécan doit être envisagée avec une prudence toute particulière, cette association pouvant majorer sa toxicité.
- La coadministration d’Emend et d’alcaloïdes dérivés de l’ergot de seigle, qui sont des substrats du CYP3A4, peut entraîner une élévation des concentrations plasmatiques de ces substances actives. Par conséquent, la prudence s’impose en raison du risque éventuel de toxicité lié à l’ergot de seigle.
- La coadministration d’Emend et de warfarine entraîne une diminution du temps de Quick, exprimé en INR (International Normalized Ratio). Chez les patients traités au long cours par la warfarine, l’INR doit être étroitement surveillé au cours du traitement par Emend et pendant les 2 semaines qui suivent chaque cure de 3 jours d’Emend pour la prévention des nausées et vomissements induits par une chimiothérapie (cf Interactions).
- L’efficacité des contraceptifs hormonaux peut être réduite pendant l’administration d’Emend et au cours des 28 jours qui suivent cette administration. Des méthodes contraceptives alternatives ou complémentaires doivent être utilisées au cours du traitement par Emend et pendant les 2 mois qui suivent la dernière prise d’Emend (cf Interactions).
- La coadministration d’Emend et de substances actives qui induisent fortement l’activité du CYP3A4 (telles que la rifampicine, la phénytoïne, la carbamazépine, le phénobarbital) doit être évitée, cette association entraînant une réduction des concentrations plasmatiques de l’aprépitant (cf Interactions). La coadministration d’Emend avec des préparations à base de plantes contenant du millepertuis (hypericum perforatum) n’est pas recommandée.
- La coadministration d’Emend et de substances actives qui inhibent l’activité du CYP3A4 (telles que le kétoconazole, l’itraconazole, le voriconazole, le posaconazole, la clarithromycine, la télithromycine, la néfazodone et les inhibiteurs de protéase) doit être envisagée avec précaution, une élévation des concentrations plasmatiques de l’aprépitant étant attendue avec cette association (cf Interactions).
- Emend contient du sucrose. Les patients ayant des problèmes héréditaires rares d’intolérance au fructose, de malabsorption du glucose-galactose, ou de déficit en sucrase-isomaltase ne doivent pas prendre ce médicament.
INTERACTIONS |
- Effet de l’aprépitant sur la pharmacocinétique d’autres substances actives :
-
- Inhibition du CYP3A4 :
- En tant qu’inhibiteur modéré du CYP3A4, l’aprépitant (125 mg/80 mg) peut entraîner une élévation des concentrations plasmatiques des substances actives administrées de façon concomitante par voie orale et qui sont métabolisées par le CYP3A4. L’exposition totale de substrats du CYP3A4 administrés par voie orale peut augmenter jusqu’à 3 fois environ au cours du traitement de 3 jours par Emend ; l’effet attendu de l’aprépitant sur les concentrations plasmatiques des substrats du CYP3A4 administrés par voie intraveineuse est moindre. Emend ne doit pas être administré de façon concomitante avec le pimozide, la terfénadine, l’astémizole ou le cisapride (cf Contre-indications). L’inhibition du CYP3A4 par l’aprépitant peut entraîner une élévation des concentrations plasmatiques de ces substances actives, susceptible de provoquer des réactions graves ou de mettre en jeu le pronostic vital. La prudence s’impose lors de la coadministration d’Emend et de substances actives administrées par voie orale, métabolisées principalement par le CYP3A4 et ayant une marge thérapeutique étroite, telles que la ciclosporine, le tacrolimus, le sirolimus, l’évérolimus, l’alfentanil, la diergotamine, l’ergotamine, le fentanyl et la quinidine (cf Mises en garde et Précautions d’emploi).
-
- Corticostéroïdes :
-
- Dexaméthasone : la dose orale habituelle de dexaméthasone doit être réduite d’environ 50 % en cas de coadministration avec Emend selon le schéma posologique de 125 mg/80 mg. La dose de dexaméthasone au cours des essais cliniques portant sur les nausées et vomissements induits par une chimiothérapie a été choisie en tenant compte des interactions entre les substances actives (cf Posologie et Mode d’administration). L’administration d’Emend 125 mg en association à une dose orale de 20 mg de dexaméthasone à J1, suivie de l’administration d’une dose de 80 mg/jour d’Emend en association à une dose orale de 8 mg de dexaméthasone de J2 à J5, a entraîné une élévation de l’ASC de la dexaméthasone, un substrat du CYP3A4, de 2,2 fois à J1 et J5.
- Méthylprednisolone : la dose habituelle de méthylprednisolone administrée par voie intraveineuse doit être réduite d’environ 25 %, et la dose orale habituelle de méthylprednisolone doit être réduite d’environ 50 % en cas de coadministration avec Emend selon le schéma posologique de 125 mg/80 mg. L’administration d’Emend selon le schéma posologique de 125 mg à J1, suivis de 80 mg/jour à J2 et J3, a augmenté l’ASC de la méthylprednisolone, substrat du CYP3A4, de 1,3 fois à J1 et de 2,5 fois à J3, lors de la coadministration de 125 mg de méthylprednisolone par voie intraveineuse à J1 et de 40 mg de méthylprednisolone par voie orale à J2 et J3.
- Au cours d’un traitement continu avec la méthylprednisolone, l’ASC de la méthylprednisolone peut diminuer au plus tard dans les 2 semaines qui suivent l’initiation du traitement par Emend, à cause de l’effet inducteur de l’aprépitant sur le CYP3A4. On peut s’attendre à ce que cet effet soit plus prononcé avec la méthylprednisolone administrée par voie orale.
- Dexaméthasone : la dose orale habituelle de dexaméthasone doit être réduite d’environ 50 % en cas de coadministration avec Emend selon le schéma posologique de 125 mg/80 mg. La dose de dexaméthasone au cours des essais cliniques portant sur les nausées et vomissements induits par une chimiothérapie a été choisie en tenant compte des interactions entre les substances actives (cf Posologie et Mode d’administration). L’administration d’Emend 125 mg en association à une dose orale de 20 mg de dexaméthasone à J1, suivie de l’administration d’une dose de 80 mg/jour d’Emend en association à une dose orale de 8 mg de dexaméthasone de J2 à J5, a entraîné une élévation de l’ASC de la dexaméthasone, un substrat du CYP3A4, de 2,2 fois à J1 et J5.
-
- Agents chimiothérapeutiques :
- Lors d’études de pharmacocinétique, l’administration d’Emend à la posologie de 125 mg à J1 suivie de 80 mg/jour à J2 et J3 n’a pas modifié la pharmacocinétique du docétaxel administré par voie intraveineuse à J1 ou de la vinorelbine administrée par voie intraveineuse à J1 ou J8. L’effet d’Emend sur la pharmacocinétique des substrats du CYP3A4 administrés par voie orale est supérieur à celui sur la pharmacocinétique des substrats du CYP3A4 administrés par voie intraveineuse ; par conséquent, une interaction avec les agents chimiothérapeutiques administrés par voie orale et métabolisés principalement ou en partie par le CYP3A4 (par exemple l’étoposide, la vinorelbine) ne peut être exclue. La prudence est recommandée et une surveillance supplémentaire peut être appropriée chez les patients recevant ce type de médicament par voie orale (cf Mises en garde et Précautions d’emploi).
-
- Immunosuppresseurs :
- Une augmentation transitoire modérée, suivie d’une légère diminution de l’exposition aux immunosuppresseurs métabolisés par le CYP3A4 (tels que la ciclosporine, le tacrolimus, l’évérolimus et le sirolimus) est attendue au cours des 3 jours de traitement administré pour la prévention des nausées et des vomissements associés à une chimiothérapie. La durée de traitement de 3 jours étant courte, les variations de l’exposition limitées et fonction du temps, aucune réduction de la dose des immunosuppresseurs n’est recommandée pendant ces 3 jours d’administration concomitante avec Emend.
-
- Midazolam :
- Les effets potentiels des concentrations plasmatiques accrues du midazolam ou d’autres benzodiazépines métabolisées par le CYP3A4 (alprazolam, triazolam) doivent être envisagés en cas de coadministration de ces médicaments et d’Emend (125 mg/80 mg).
- Emend a augmenté l’ASC du midazolam, substrat sensible du CYP3A4, de 2,3 fois à J1 et de 3,3 fois à J5, lorsqu’une dose orale unique de 2 mg de midazolam a été associée à J1 et à J5 au schéma posologique de 125 mg d’Emend à J1 suivis de 80 mg/jour de J2 à J5.
- Dans une autre étude avec administration intraveineuse de midazolam, Emend a été administré à la posologie de 125 mg à J1 suivis de 80 mg/jour à J2 et J3, et 2 mg de midazolam ont été administrés par voie intraveineuse avant l’administration d’Emend selon le schéma posologique de 3 jours ainsi qu’à J4, J8 et J15. Emend a augmenté l’ASC du midazolam de 25 % à J4 et a diminué l’ASC du midazolam de 19 % à J8 et de 4 % à J15. Ces effets n’ont pas été considérés comme cliniquement importants.
- Dans une troisième étude avec administration intraveineuse et orale de midazolam, Emend a été administré à la posologie de 125 mg à J1 suivis de 80 mg/jour à J2 et J3, associé à 32 mg d’ondansétron à J1, à 12 mg de dexaméthasone à J1 et 8 mg de dexaméthasone de J2 à J4. Cette association (c’est-à-dire Emend, ondansétron et dexaméthasone) a diminué l’ASC du midazolam administré par voie orale de 16 % à J6, 9 % à J8, 7 % à J15 et 17 % à J22. Ces effets n’ont pas été considérés comme cliniquement importants.
- Une étude supplémentaire a été réalisée avec administration intraveineuse de midazolam et d’Emend. 2 mg de midazolam ont été administrés par voie intraveineuse 1 heure après une prise unique d’Emend 125 mg par voie orale. L’ASC plasmatique du midazolam a été augmentée de 1,5 fois. Cet effet n’a pas été considéré comme cliniquement important.
-
- Induction :
- En tant qu’inducteur léger du CYP2C9, du CYP3A4 et de la glucuronidation, l’aprépitant peut diminuer les concentrations plasmatiques des substrats éliminés par ces voies. Cet effet peut n’apparaître qu’après la fin du traitement par Emend. Pour les substrats du CYP2C9 et du CYP3A4, l’induction est transitoire avec un effet maximal atteint 3 à 5 jours après la fin du traitement de 3 jours par Emend. L’effet persiste pendant quelques jours, diminue ensuite lentement et est cliniquement non significatif 2 semaines après la fin du traitement par Emend. Une induction légère de la glucuronidation est également constatée avec 80 mg d’aprépitant administrés par voie orale pendant 7 jours. Il n’y a pas de données concernant les effets sur le CYP2C8 et le CYP2C19. La prudence s’impose lors de l’administration pendant cette période de warfarine, d’acénocoumarol, de tolbutamide, de phénytoïne ou d’autres substances actives connues pour être métabolisées par le CYP2C9.
-
- Warfarine :
- Chez les patients sous traitement chronique par la warfarine, le temps de Quick (INR) doit être surveillé étroitement au cours du traitement par Emend et au cours des 2 semaines suivant chaque cure de 3 jours d’Emend pour la prévention des nausées et vomissements induits par une chimiothérapie (cf Mises en garde et Précautions d’emploi). Lors de l’administration d’une prise unique de 125 mg d’Emend à J1, suivie de celle de 80 mg/jour à J2 et J3, à des sujets sains stabilisés traités au long cours par la warfarine, il n’y a pas eu d’effet d’Emend sur l’ASC plasmatique de la R(+) ou de la S(-) warfarine à J3 ; cependant, il y a eu une réduction de 34 % de la concentration résiduelle de la S(-) warfarine (un substrat du CYP2C9), accompagnée d’une diminution de 14 % de l’INR, 5 jours après la fin du traitement par Emend.
-
- Tolbutamide :
- Emend, administré à la dose de 125 mg à J1 et de 80 mg/jour à J2 et J3, a abaissé l’ASC du tolbutamide (un substrat du CYP2C9) de 23 % à J4, de 28 % à J8 et de 15 % à J15, lors de l’administration d’une prise orale unique de 500 mg de tolbutamide avant l’administration d’Emend selon le schéma posologique de 3 jours à J4, J8 et J15.
-
- Contraceptifs hormonaux :
- L’efficacité des contraceptifs hormonaux peut être réduite pendant l’administration d’Emend et au cours des 28 jours qui la suivent. Des méthodes contraceptives alternatives ou complémentaires doivent être utilisées au cours du traitement par Emend et pendant les 2 mois qui suivent la dernière prise d’Emend.
- Dans une étude clinique, une prise unique d’un contraceptif oral contenant de l’éthinylestradiol et de la noréthindrone a été administrée de J1 à J21 avec Emend pris selon le schéma posologique de 125 mg à J8 puis 80 mg/jour à J9 et J10, associé à 32 mg d’ondansétron par voie intraveineuse à J8 et à la dexaméthasone par voie orale à la posologie de 12 mg à J8 et 8 mg/jour à J9, J10 et J11. Dans cette étude, de J9 à J21, il y a eu une diminution allant jusqu’à 64 % des concentrations résiduelles d’éthinylestradiol et une diminution allant jusqu’à 60 % des concentrations résiduelles de noréthindrone.
-
- Antagonistes 5-HT3 :
- Au cours des études cliniques d’interaction, l’aprépitant n’a pas eu d’effet cliniquement important sur la pharmacocinétique de l’ondansétron, du granisétron ou de l’hydrodolasétron (le métabolite actif du dolasétron).
- Effet d’autres médicaments sur la pharmacocinétique de l’aprépitant :
- La coadministration d’Emend et de substances actives inhibant l’activité du CYP3A4 (telles que le kétoconazole, l’itraconazole, le voriconazole, le posaconazole, la clarithromycine, la télithromycine, la néfazodone et les inhibiteurs de protéase) doit être envisagée avec précaution ; une augmentation des concentrations plasmatiques d’aprépitant est attendue avec cette association (cf Mises en garde et Précautions d’emploi).
- La coadministration d’Emend et de substances actives induisant fortement l’activité du CYP3A4 (telles que la rifampicine, la phénytoïne, la carbamazépine, le phénobarbital) doit être évitée, une telle association entraînant une diminution des concentrations plasmatiques d’aprépitant, et donc une diminution de l’efficacité d’Emend. La coadministration d’Emend et de préparation à base de plantes contenant du millepertuis (hypericum perforatum) n’est pas recommandée.
-
- Kétoconazole :
- Lorsqu’une prise unique de 125 mg d’aprépitant a été administrée à J5 d’un schéma posologique de 10 jours de 400 mg/jour de kétoconazole, un puissant inhibiteur du CYP3A4, l’ASC de l’aprépitant a augmenté d’environ 5 fois et la demi-vie terminale moyenne de l’aprépitant a augmenté d’environ 3 fois.
-
- Rifampicine :
- Lorsqu’une prise unique de 375 mg d’aprépitant a été administrée à J9 d’un schéma posologique de 14 jours de 600 mg/jour de rifampicine, un puissant inducteur du CYP3A4, l’ASC de l’aprépitant a diminué de 91 % et la demi-vie terminale moyenne a diminué de 68 %.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
| Investigations : | |
| Fréquent | Élévation des ALAT, élévation des ASAT |
| Peu fréquent | Élévation des phosphatases alcalines, hyperglycémie, hématurie microscopique, hyponatrémie, perte de poids, diminution du nombre de neutrophiles |
| Affections cardiaques : | |
| Peu fréquent | Bradycardie, palpitations, troubles cardiovasculaires |
| Affections hématologiques et du système lymphatique : | |
| Peu fréquent | Neutropénie fébrile, anémie |
| Affections du système nerveux : | |
| Fréquent | Céphalées, étourdissements |
| Peu fréquent | Anomalies du rêve, troubles cognitifs, léthargie, somnolence |
| Affections oculaires : | |
| Peu fréquent | Conjonctivite |
| Affections de l’oreille et du labyrinthe : | |
| Peu fréquent | Acouphènes |
| Affections respiratoires, thoraciques et médiastinales : | |
| Fréquent | Hoquet |
| Peu fréquent | Pharyngite, éternuement, toux, écoulement rétronasal, irritation de la gorge |
| Affections gastro-intestinales : | |
| Fréquent | Constipation, diarrhée, dyspepsie, éructation |
| Peu fréquent | Ulcère duodénal perforant, nausées*, vomissements*, régurgitation acide, dysgueusie, gêne épigastrique, constipation opiniâtre, reflux gastro-oesophagien, douleur abdominale, bouche sèche, entérocolite, flatulence, stomatite, distension abdominale, selles dures, colite neutropénique |
| Affections du rein et des voies urinaires : | |
| Peu fréquent | Polyurie, dysurie, pollakiurie |
| Affections de la peau et du tissu sous-cutané : | |
| Peu fréquent | Rash, acné, photosensibilité, hyperhydrose, peau grasse, prurit, lésions cutanées, rash pruritique |
| Affections musculosquelettiques et systémiques : | |
| Peu fréquent | Crampes musculaires, myalgies, faiblesse musculaire |
| Troubles du métabolisme et de la nutrition : | |
| Fréquent | Anorexie |
| Peu fréquent | Prise de poids, polydipsie |
| Infections et infestations : | |
| Peu fréquent | Candidose, infection à staphylocoques |
| Affections vasculaires : | |
| Peu fréquent | Bouffées vasomotrices/bouffées de chaleur |
| Troubles généraux et anomalies au site d’administration : | |
| Fréquent | Asthénie/fatigue |
| Peu fréquent | OEdème, gêne thoracique, malaise, soif, frissons, modifications de la démarche |
| Affections psychiatriques : | |
| Peu fréquent | Désorientation, euphorie, anxiété |
* Les nausées et vomissements étaient des paramètres d’efficacité au cours des 5 premiers jours suivant la chimiothérapie et n’étaient rapportés comme événements indésirables qu’ensuite.
- Depuis la commercialisation :
- Les effets indésirables suivants ont été rapportés depuis la mise sur le marché (fréquence indéterminée) :
- Affections de la peau et du tissu sous-cutané : prurit, éruption, urticaire.
- Affections du système immunitaire : réactions d’hypersensibilité incluant des réactions anaphylactiques.
- Affections de la peau et du tissu sous-cutané : prurit, éruption, urticaire.
SURDOSAGE |
Somnolence et céphalées ont été rapportées chez un patient ayant ingéré 1440 mg d’aprépitant.
L’aprépitant ne peut être éliminé par hémodialyse.
PHARMACODYNAMIE |
Groupe pharmacothérapeutique : antiémétiques et antinauséeux (code ATC : A04AD12).
L’aprépitant est un antagoniste sélectif à haute affinité pour les récepteurs de la substance P neurokinine 1 (NK1) humaine.
Au cours de deux études randomisées en double aveugle incluant un total de 1094 patients sous chimiothérapie, avec une dose de cisplatine >= 70 mg/m2, l’aprépitant en association à un schéma posologique ondansétron/dexaméthasone (cf Posologie et Mode d’administration) a été comparé à un schéma posologique standard (placebo + 32 mg d’ondansétron administrés en intraveineux à J1 + 20 mg de dexaméthasone par voie orale à J1 et 8 mg par voie orale 2 fois par jour de J2 à J4).
L’efficacité a été évaluée sur la base du critère composite suivant : réponse complète (définie par l’absence d’épisodes émétiques et l’absence de recours à un traitement de secours), principalement au cours du cycle 1. Les résultats ont été évalués individuellement pour chaque étude ainsi que pour les deux études combinées.
Un résumé des résultats clés issus de l’analyse combinée des études est donné dans le tableau 1.
| Critères composites | Aprépitant (N = 521)* % | Traitement standard (N = 524)* % | Différences**
% (IC 95 %) |
| Réponse complète
(pas de vomissements ; pas de traitement de secours) |
|||
| Total (0-120 h) | 67,7 | 47,8 | 19,9 (14,0 ; 25,8) |
| 0 – 24 h | 86,0 | 73,2 | 12,7 (7,9 ; 17,6) |
| 25 – 120 h | 71,5 | 51,2 | 20,3 (14,5 ; 26,1) |
| Critères individuels | Aprépitant (N = 521)* % | Traitement standard (N = 524)* % | Différences**
% (IC 95 %) |
| Pas de vomissements (pas d’épisodes émétiques
avec ou sans traitement de secours) |
|||
| Total (0 – 120 h) | 71,9 | 49,7 | 22,2 (16,4 ; 28,0) |
| 0 – 24 h | 86,8 | 74,0 | 12,7 (8,0 ; 17,5) |
| 25 – 120 h | 76,2 | 53,5 | 22,6 (17,0 ; 28,2) |
| Pas de nausées significatives
(VAS max < 25 mm sur une échelle de 0 à 100 mm) |
|||
| Total (0 – 120 h) | 72,1 | 64,9 | 7,2 (1,6 ; 12,8) |
| 25 – 120 h | 74,0 | 66,9 | 7,1 (1,5 ; 12,6) |
* Un patient dans le groupe aprépitant a été exclu de l’analyse globale et de celle de la phase retardée, ses données n’étant disponibles que pour la phase aiguë ; un patient recevant le traitement standard a été exclu de l’analyse globale et de celle de la phase aiguë, ses données n’étant disponibles que pour la phase retardée.
** Les intervalles de confiance ont été calculés sans ajustement en fonction du sexe et des chimiothérapies concomitantes, lesquels avaient été pris en compte dans l’analyse primaire du risque relatif et des modèles logistiques.
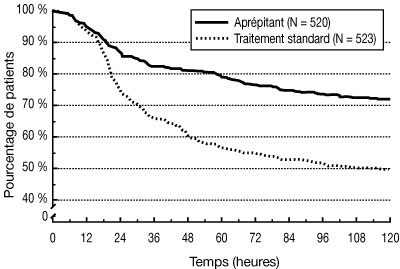
Dans l’analyse combinée, le temps estimé jusqu’au premier vomissement est donné par la courbe de Kaplan-Meier sur la figure 1.
| Figure 1 : Pourcentage de patients recevant une chimiothérapie hautement émétisante et indemnes de vomissements – cycle 1 : |
 |
Des différences statistiquement significatives dans l’efficacité ont également été observées individuellement dans chacune des deux études.
Dans le cadre de ces deux mêmes études cliniques, 851 patients ont poursuivi une extension de l’évaluation lors des cycles ultérieurs allant jusqu’à 5 cycles supplémentaires de chimiothérapie. L’efficacité du schéma aprépitant s’est apparemment maintenue durant tous les cycles.
Dans une étude randomisée, en double aveugle, réalisée sur un total de 866 patients (864 femmes, 2 hommes) recevant une chimiothérapie comprenant soit du cyclophosphamide 750-1500 mg/m2, soit du cyclophosphamide 500-1500 mg/m2 et de la doxorubicine (<= 60 mg/m2) ou de l’épirubicine (<= 100 mg/m2), l’aprépitant en association à un traitement ondansétron/dexaméthasone (cf Posologie et Mode d’administration) a été comparé à un traitement standard (placebo + 8 mg d’ondansétron par voie orale [2 fois à J1 et toutes les 12 heures à J2 et J3] + 20 mg de dexaméthasone 20 mg par voie orale à J1).
L’efficacité a été évaluée sur la base du critère composite : réponse complète (définie par l’absence d’épisodes émétiques et l’absence de recours à un traitement de secours), principalement au cours du cycle 1.
Un résumé des résultats clés de l’étude est donné dans le tableau 2.
| Critères composites | Aprépitant (N = 433)* % | Traitement standard (N = 424) % | Différences**
% (IC 95 %) |
| Réponse complète
(pas de vomissements ; pas de traitement de secours) |
|||
| Total (0 – 120 h) | 50,8 | 42,5 | 8,3 (1,6 ; 15,0) |
| 0 – 24 h | 75,7 | 69,0 | 6,7 (0,7 ; 12,7) |
| 25 – 120 h | 55,4 | 49,1 | 6,3 (- 0,4 ; 13,0) |
| Critères individuels | Aprépitant (N = 433)* % | Traitement standard (N = 424) % | Différences**
% (IC 95 %) |
| Pas de vomissements (pas d’épisodes émétiques
avec ou sans traitement de secours) |
|||
| Total (0 – 120 h) | 75,7 | 58,7 | 17,0 (10,8 ; 23,2) |
| 0 – 24 h | 87,5 | 77,3 | 10,2 (5,1 ; 15,3) |
| 25 – 120 h | 80,8 | 69,1 | 11,7 (5,9 ; 17,5) |
| Pas de nausées significatives (VAS max < 25 mm sur une échelle de 0 à 100 mm) |
|||
| Total (0 – 120 h) | 60,9 | 55,7 | 5,3 (- 1,3 ; 11,9) |
| 0 – 24 h | 79,5 | 78,3 | 1,3 (- 4,2 ; 6,8) |
| 25 – 120 h | 65,3 | 61,5 | 3,9 (- 2,6 ; 10,3) |
* Un patient dans le groupe aprépitant a été exclu de l’analyse globale et de celle de la phase retardée, ses données n’étant disponibles que pour la phase aiguë.
** Les intervalles de confiance ont été calculés sans ajustement en fonction de la tranche d’âge (< 55 ans, >= 55 ans) et du groupe d’investigateur, lesquels avaient été pris en compte dans l’analyse primaire du risque relatif et des modèles logistiques.
Dans le cadre de cette même étude clinique, 744 patients ont poursuivi une extension de l’évaluation lors des cycles ultérieurs allant jusqu’à 3 cycles supplémentaires de chimiothérapie. L’efficacité du schéma aprépitant s’est apparemment maintenue durant tous les cycles.
Dans une seconde étude clinique multicentrique, randomisée, en double aveugle, sur des groupes parallèles, l’aprépitant a été comparé au traitement standard chez 848 patients (652 femmes, 196 hommes) recevant une chimiothérapie qui comportait une administration IV d’oxaliplatine, de carboplatine, d’épirubicine, d’idarubicine, d’ifosfamide, d’irinotécan, de daunorubicine, de doxorubicine, ou du cyclophosphamide IV (< 1 500 mg/m2) ou de la cytarabicine IV (> 1 g/m2). Les patients sous aprépitant recevaient une chimiothérapie pour divers types de tumeurs dont 52 % de cancers du sein, 21 % de cancers gastro-intestinaux y compris le cancer colorectal, 13 % de cancers pulmonaires et 6 % de cancers gynécologiques. L’aprépitant en association à un traitement ondansétron/dexaméthasone (cf Posologie et Mode d’administration) a été comparé au traitement standard (placebo associé à 8 mg d’ondansétron par voie orale [2 fois à J1 et toutes les 12 heures à J2 et J3] plus 20 mg de dexaméthasone par voie orale à J1).
L’efficacité était basée sur l’évaluation du critère primaire et du principal critère secondaire suivants : pas de vomissements pendant toute la période (de 0 à 120 heures après la chimiothérapie), évaluation de la sécurité d’emploi et de la tolérance de l’aprépitant pour le traitement des nausées et vomissements induits par une chimiothérapie ainsi que la réponse complète (pas de vomissements et pas de traitement de secours) pendant toute la période (0 à 120 heures après la chimiothérapie). De plus, le critère « Pas de nausées significatives pendant toute la période (0-120 h après la chimiothérapie) » a été évalué à titre exploratoire et dans les phases aiguë et retardée sous forme d’analyse post-hoc.
Un résumé des résultats clés de l’étude est donné dans le tableau 3.
| Aprépitant (N = 425) % | Traitement standard (N = 406) % | Différences(1)
% (IC 95 %) |
|
| Réponse complète (pas de vomissements et pas de traitement de secours) | |||
| Total (0-120 h) | 68,7 | 56,3 | 12,4 (5,9 ; 18,9) |
| 0 – 24 h | 89,2 | 80,3 | 8,9 (4,0 ; 13,8) |
| 25 – 120 h | 70,8 | 60,9 | 9,9 (3,5 ; 16,3) |
| Pas de vomissements (pas d’épisodes émétiques avec ou sans traitement de secours) | |||
| Total (0 – 120 h) | 76,2 | 62,1 | 14,1 (7,9 ; 20,3) |
| 0 – 24 h | 92,0 | 83,7 | 8,3 (3,9 ; 12,7) |
| 25 – 120 h | 77,9 | 66,8 | 11,1 (5,1 ; 17,1) |
| Pas de nausées significatives
(VAS max < 25 mm sur une échelle de 0 à 100 mm) |
|||
| Total (0 – 120 h) | 73,6 | 66,4 | 7,2 (1,0 ; 13,4) |
| 0 – 24 h | 90,9 | 86,3 | 4,6 (0,2 ; 9,0) |
| 25 – 120 h | 74,9 | 69,5 | 5,4 (- 0,7 ; 11,5) |
(1) Les intervalles de confiance ont été calculés sans ajustement en fonction du sexe et de la localisation de la tumeur, lesquels avaient été pris en compte dans l’analyse primaire utilisant des modèles logistiques.
Le bénéfice du traitement par l’aprépitant associé au traitement standard dans la population totale de l’étude est principalement dû aux résultats observés chez les patients faiblement contrôlés par le traitement standard tels que les femmes, même si les résultats sont supérieurs en nombre quels que soient l’âge, le type de tumeur ou le sexe. La réponse complète à l’aprépitant et au traitement standard a été atteinte chez respectivement 209/324 (65 %) et 161/320 (50 %) des femmes et chez 83/101 (82 %) et 68/87 (78 %) des hommes.
PHARMACOCINÉTIQUE |
L’aprépitant présente une pharmacocinétique non linéaire. La clairance et la biodisponibilité absolue diminuent toutes deux avec l’augmentation de la dose.
- Absorption :
- La biodisponibilité absolue moyenne de l’aprépitant par voie orale est de 67 % pour la gélule de 80 mg et de 59 % pour la gélule de 125 mg. Le pic moyen de concentration plasmatique (Cmax) de l’aprépitant est survenu aux environs de la 4e heure (Tmax). L’administration de la gélule avec un petit déjeuner standard d’environ 800 kcal a entraîné une augmentation de 40 % de l’ASC de l’aprépitant. Cette augmentation n’est pas jugée pertinente sur le plan clinique.
- La pharmacocinétique de l’aprépitant est non linéaire sur l’éventail des doses cliniques. Chez le jeune adulte sain, l’augmentation de l’ASC0-infini a été de 26 % supérieure à la proportionnalité de la dose, pour des doses uniques de 80 et de 125 mg administrées non à jeun.
- Après administration orale d’une dose unique de 125 mg d’Emend à J1 et de 80 mg une fois par jour à J2 et J3, l’ASC0-24 h (moyenne ± ET) a été de 19,6 µg × h/ml ± 2,5 et de 21,2 µg × h/ml ± 6,3 à J1 et J3 respectivement. La Cmax a été de 1,6 µg/ml ± 0,36 et de 1,4 µg/ml ± 0,22 à J1 et J3 respectivement.
- Distribution :
- L’aprépitant se lie fortement aux protéines, avec une moyenne de 97 %. La moyenne géométrique du volume apparent de distribution à l’état d’équilibre (Vdss) est d’environ 66 l chez l’homme.
- Métabolisme :
- L’aprépitant subit un métabolisme important. Chez le jeune adulte sain, l’aprépitant représente environ 19 % de la radioactivité mesurée au niveau du plasma durant les 72 heures qui suivent l’administration d’une dose intraveineuse unique de 100 mg de fosaprépitant marquée au [14C], une prodrogue de l’aprépitant, ce qui indique une présence substantielle de métabolites au niveau du plasma. Douze métabolites de l’aprépitant ont été identifiés dans le plasma humain. Le métabolisme de l’aprépitant intervient largement via l’oxydation au niveau du cycle de la morpholine et de ses chaînes latérales, les métabolites qui en résultent n’étant que faiblement actifs. Les études réalisées in vitro sur des microsomes hépatiques humains indiquent que l’aprépitant est tout d’abord métabolisé au niveau du CYP3A4, et potentiellement dans une moindre proportion par les CYP1A2 et CYP2C19.
- Élimination :
- L’aprépitant n’est pas excrété sous forme inchangée dans les urines. Les métabolites sont excrétés dans les urines et, par voie biliaire, dans les fèces. Après administration à des sujets sains d’une dose intraveineuse unique de 100 mg de fosaprépitant, une prodrogue de l’aprépitant marquée au [14C], 57 % de la radioactivité ont été récupérés dans les urines et 45 % dans les fèces.
- La clairance plasmatique de l’aprépitant est dose-dépendante et décroît avec l’augmentation de la dose, allant de 60 à 72 ml/min environ dans la fourchette des doses thérapeutiques. La demi-vie terminale va de 9 à 13 heures environ.
- Pharmacocinétique chez des populations particulières :
-
- Sujet âgé : après administration orale d’une dose unique de 125 mg d’aprépitant à J1 et de 80 mg une fois par jour de J2 à J5, l’ASC0-24 h de l’aprépitant a été de 21 % supérieure à J1 et de 36 % à J5 chez le sujet âgé (>= 65 ans), comparée au jeune adulte. La Cmax a été supérieure de 10 % à J1 et de 24 % à J5 chez le sujet âgé, comparée au jeune adulte. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement posologique d’Emend n’est nécessaire chez les patients âgés.
- Sexe : après administration orale d’une dose unique de 125 mg d’aprépitant, la Cmax de l’aprépitant a été supérieure de 16 % chez les femmes, comparée aux hommes. La demi-vie de l’aprépitant a été inférieure de 25 % chez les femmes, comparée aux hommes, alors que le Tmax survient approximativement au même moment. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement posologique d’Emend n’est nécessaire en fonction du sexe.
- Insuffisance hépatique : une insuffisance hépatique légère (classe A de Child-Pugh) n’affecte pas la pharmacocinétique de l’aprépitant de façon cliniquement significative. Aucun ajustement posologique n’est nécessaire chez les patients en insuffisance hépatique légère. On ne peut pas tirer de conclusions concernant l’influence d’une insuffisance hépatique modérée (classe B de Child-Pugh) sur la pharmacocinétique de l’aprépitant à partir des données actuellement disponibles. On ne dispose d’aucune donnée clinique ni pharmacocinétique chez les patients en insuffisance hépatique sévère (classe C de Child-Pugh).
- Insuffisance rénale : une dose unique de 240 mg d’aprépitant a été administrée à des patients en insuffisance rénale sévère (Clcr < 30 ml/min) et à des patients atteints de néphropathie à un stade terminal nécessitant une hémodialyse. Chez les patients en insuffisance rénale sévère, l’ASC0-infini de l’aprépitant total (lié ou non aux protéines) a diminué de 21 % et la Cmax a diminué de 32 % comparé à des sujets sains. Chez les patients atteints de néphropathie à un stade terminal et sous hémodialyse, l’ASC0-infini de l’aprépitant total a diminué de 42 % et la Cmax a diminué de 32 %. En raison d’une baisse modeste de la liaison protéique de l’aprépitant chez les patients atteints de néphropathie, l’ASC de la molécule non liée et pharmacologiquement active n’est pas affectée de façon significative chez les patients insuffisants rénaux comparés aux sujets sains. Une hémodialyse réalisée entre 4 et 48 heures après la prise n’a eu aucun effet significatif sur la pharmacocinétique de l’aprépitant ; moins de 0,2 % de la dose a été récupérée au niveau du dialysat. Aucun ajustement posologique d’Emend n’est nécessaire chez les patients en insuffisance rénale ni chez les patients atteints d’une néphropathie à un stade terminal sous hémodialyse.
- Relation effet/dose : à l’aide d’un traceur hautement spécifique du récepteur NK1, des études par tomographie par émission de positrons (TEP), menées auprès de jeunes hommes sains, ont montré que l’aprépitant pénètre dans le cerveau et se lie aux récepteurs NK1 de façon dose et concentration plasmatique dépendantes. Les concentrations plasmatiques de l’aprépitant obtenues selon le schéma posologique de 3 jours d’Emend permettent d’envisager un taux de liaison aux récepteurs cérébraux NK1 supérieur à 95 %.
- Sujet âgé : après administration orale d’une dose unique de 125 mg d’aprépitant à J1 et de 80 mg une fois par jour de J2 à J5, l’ASC0-24 h de l’aprépitant a été de 21 % supérieure à J1 et de 36 % à J5 chez le sujet âgé (>= 65 ans), comparée au jeune adulte. La Cmax a été supérieure de 10 % à J1 et de 24 % à J5 chez le sujet âgé, comparée au jeune adulte. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement posologique d’Emend n’est nécessaire chez les patients âgés.
SÉCURITE PRÉCLINIQUE |
Les données précliniques ne révèlent aucun risque particulier pour l’homme, sur la base des études conventionnelles de toxicité à doses unique et multiple, de génotoxicité, du potentiel cancérigène, et de toxicité sur la fonction de reproduction. Toutefois, il convient de noter que l’exposition systémique chez des rongeurs a été comparable, voire inférieure, à l’exposition chez l’homme à des doses thérapeutiques de 125 mg/80 mg. En particulier, bien qu’aucun effet indésirable n’ait été noté lors des études sur la fonction de reproduction à des niveaux d’exposition humaine, les expositions chez l’animal n’ont pas été suffisantes pour permettre une évaluation adéquate du risque chez l’homme.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 4 ans.
A conserver dans l’emballage extérieur d’origine, à l’abri de l’humidité.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM | EU/1/03/262/002 ; CIP 3400936350700 (RCP rév 01.02.2011) 2 gél à 80 mg. |
| EU/1/03/262/003 ; CIP 3400956493081 (RCP rév 01.02.2011) 5 gél à 80 mg. | |
| EU/1/03/262/005 ; CIP 3400956493142 (RCP rév 01.02.2011) 5 gél à 125 mg. | |
| EU/1/03/262/006 ; CIP 3400936351189 (RCP rév 01.02.2011) 1 gél à 125 mg et 2 gél à 80 mg. |
| Prix : | 52.60 euros (2 gélules à 80 mg). |
| 76.29 euros (1 gélule à 125 mg et 2 gélules à 80 mg). | |
|
Remb Séc soc à 65 % selon la procédure des médicaments d’exception (prescription en conformité avec la fiche d’information thérapeutique). Collect. |
|
| Modèles hospitaliers : Collect. | |
Laboratoires MERCK SHARP & DOHME-CHIBRET
3, av Hoche. 75114 Paris cdx 08
Tél : 01 47 54 87 00
Info médic : Tél : 01 47 54 88 00
Site web : http://www.msd-france.com
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale


